
Novel rhodanines with anticancer activity: design, synthesis and CoMSIA study
Subhankar P. Mandala,
Mithunaa,
Aakriti Garga,
Sanjana S. Sahetyaa,
S. R. Nagendraa,
H. S. Sripada,
Mendon Manisha Manjunatha,
Sitarama,
Mukesh Sonia,
R. Nasir Baigb,
S. Vasanth Kumarc and
B. R. Prashantha Kumar*a
aDepartment of Pharmaceutical Chemistry, JSS College of Pharmacy, Mysuru 570 015, India. E-mail: brprashanthkumar@jssuni.edu.in; Fax: +91-821-2548359; Tel: +91-821-2548353
bDepartment of Organic Chemistry, Indian Institute of Science, Bengaluru 560 012, India
cDepartment of Mathematics, National Institute of Engineering, Mysuru 570 008, India
First published on 14th June 2016
Abstract
Three different series of some novel N-substituted rhodanines were designed for anticancer activity and prepared from the corresponding dithiocarbamates. The synthesized compounds were analyzed by IR, NMR and MASS to confirm their structures. All the titled compounds were found to be of Z configuration based on NMR spectral analysis. All the synthesized rhodanines were screened for in vitro anticancer activity against MCF-7 breast cancer cells at the concentration of 10 μg. The compounds showed moderate to significant cytotoxicity. Amongst them, interestingly, compounds 10, 22 and 33 with cinnamoyl substitution at the 5th position of the thiazolidine ring system showed significant activity. Further, we subjected all these compounds to a CoMSIA study to study their 3D quantitative structure activity relationships (3D QSAR). The illustration about the design of novel rhodanines, synthesis, analysis, activity against MCF-7 cells and SAR via CoMSIA study are reported here.
1. Introduction
Cancer is one of the second leading causes of death across the world. The incidence and burden of cancer is huge and is set to rise.1–3 Cancer kills more people on a global scale than AIDS, malaria and tuberculosis combined. Among the different types of cancers reported so far breast cancer is the second major reason for deaths among women.4 Apart from the use of surgery and radiotherapy, chemotherapy still remains an important option for the treatment of cancer. Despite the many efforts to implement novel chemotherapeutic strategies for the treatment of different types of cancer, this disease still remains one of the major concerns worldwide. As a result, a need of the hour in this area of drug research is to find a new class of molecules with selective action against cancer cells.In search of a potent anticancer agent, an effort has been made in the recent past to develop some novel molecules containing various heterocyclic scaffolds. In this context, five-membered heterocyclic molecules containing thiazolidine nucleus with a carbonyl group on fourth carbon such as rhodanine and its bioisostere 2,4-thiazolidinedione (TZD) derivatives have exhibited a broad spectrum of anticancer activity.5–13
TZD's and rhodanines are known to have biological activities, such as-anti-diabetic,14 anti-inflammatory,15 anti-oxidant,16 anti-tubercular,17 anti-microbial,17,18 anticonvulsant19 and cytotoxic activities.20 The pyridinylquinoline derivative GSK1059615 (Fig. 1) is a novel, ATP-competitive, and reversible inhibitor of the class I family of PI3Ks. It inhibits phosphatidylinositol 3-kinase (PI3K) signaling, induces G1 arrest and apoptosis, especially in breast tumor cells.21,22 Moorthy et al., have reported 5-benzilidene-3-ethyl rhodanine (BTR-1) (Fig. 1), 3-dimethyl-2-thio-hydantoin (ITH-1), 3-ethyl-2-thio-2,4-oxazolidinedione (ITO-1) and found that all the compounds induced cytotoxicity in a time and concentration-dependent manner with an IC50 value of <10 μM and affected cell division by inducing a block at S phase, which finally led to the activation of apoptosis.23 However, despite these advances, a highly active therapeutic compounds from this class is yet to be explored for the treatment of cancer.24,25
 | ||
| Fig. 1 Rhodanines with potential anticancer activity. | ||
In addition to the above, TZDs are implicated in cancer development, progression, and metastasis, among which the Raf/MEK/extracellular signal-regulated kinase (ERK),26 PI3K,26 Wnt signal transduction pathways27 and peroxisome proliferator-activated receptors28,29 signaling cascades are the most commonly up-regulated in human cancers. This is the reason why investigation/molecular modification and pharmacological evaluation of rhodanines or TZDs have attracted special attention of synthetic chemists and pharmacologists, respectively.30 Therefore, an attempt has been made in the present study to design, synthesize and screen a series of novel rhodanines for their anticancer activity.
2. Results and discussion
2.1. Chemistry
Considering the structures of GSK1059615 and BTR-I it is evident that, the five-membered 1,3-thiazolidine ring is the basic scaffold. The presence of carbonyl groups at the second and fourth position seems to be essential for the activity. It seems that, the carbonyl group at second position of TZD can be replaced with its bioisostere C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S to form a rhodanine ring. The substitution with hydrophobic groups at the third and fifth position appears to be essential for the anticancer activity. Considering these findings, here, we designed some novel rhodanines with varied substitutions at third and fifth positions of rhodanine ring system. The synthetic scheme followed to synthesize the designed library of compounds is as outlined in Scheme 1.
S to form a rhodanine ring. The substitution with hydrophobic groups at the third and fifth position appears to be essential for the anticancer activity. Considering these findings, here, we designed some novel rhodanines with varied substitutions at third and fifth positions of rhodanine ring system. The synthetic scheme followed to synthesize the designed library of compounds is as outlined in Scheme 1.
 | ||
| Scheme 1 Synthesis of 2-thioxo-thiazolidin-4-one (rhodanine) derivatives: [a] carbon disulphide, ammonia, 0 °C, stir for 1 h [b] 5 to 10 °C, sodium chloroacetate, stir for 1 h [c] hydrochloric acid, 80 to 85 °C for 15 min [d] toluene, aldehyde, piperidine, glacial acetic acid, activated molecular sieves, reflux for 15 h. | ||
In the present work, we have selected three different amines which includes aromatic and aliphatic amines such as 2-chloroaniline, cyclohexamine and benzylamine as building blocks. These amines are first converted to their respective dithiocarbamates (1, 2 and 3) by reacting with carbon disulfide and ammonia under cold conditions. The formed dithiocarbamates then reacted with sodium chloroacetate to form N-substituted intermediates. Then these intermediates were cyclized under acidic conditions to form N-substituted rhodanines (4, 5 and 6). Finally, these rhodanines condensed with different aldehydes by the Knoevenagel condensation31 to afford the rhodanines (7–39) with different substitutions at third and fifth positions as listed in the Table 1. The present method is a simple and easy to perform. We observed that the dithiocarbamates are relatively more stable under cold conditions and there is no necessity to separate and purify. The Knoevenagel condensation was performed with relative ease using activated molecular sieves rather Dean–Stark apparatus to trap the water molecules. Alternatively, Knoevenagel condensation can be performed using microwaves to enhance reaction rates, however, yields were found to be more or less the same.
| CPD No. | R1 | % cytotoxicity | ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Cyt Cyt |
Predicted activity | Residual activity |
|---|---|---|---|---|---|
| 7 |  |
62 | 4.127 | 4.139 | −0.012 |
| 8 |  |
43 | 3.761 | 3.771 | −0.01 |
| 9 | 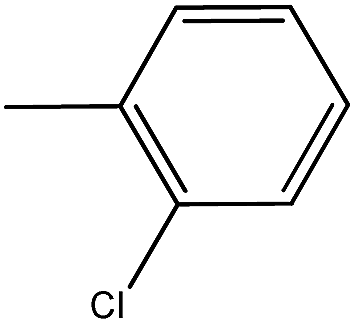 |
34 | 3.526 | 3.673 | −0.147 |
| 10 |  |
81 | 4.393 | 4.309 | 0.084 |
| 11 |  |
47 | 3.85 | 3.806 | 0.044 |
| 13 |  |
45 | 3.806 | 3.781 | 0.025 |
| 14 |  |
40 | 3.688 | 3.661 | 0.027 |
| 15 |  |
38 | 3.637 | 3.639 | −0.002 |
| 16 |  |
23 | 3.135 | 3.164 | −0.029 |
| 17 | 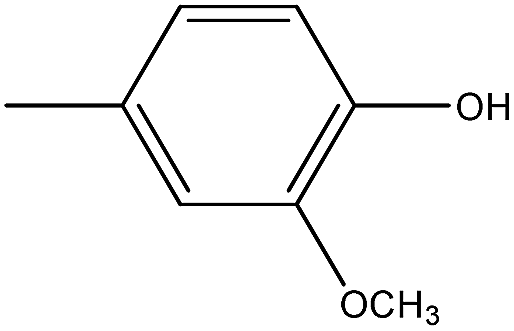 |
61 | 4.110 | 4.107 | 0.003 |
| 18 |  |
55 | 4.007 | 4.04 | −0.033 |
| 19 |  |
58 | 4.060 | 4.045 | 0.015 |
| 20 |  |
39 | 3.663 | 3.704 | −0.041 |
| 22 |  |
77 | 4.343 | 4.37 | −0.027 |
| 23 |  |
34 | 3.526 | 3.474 | 0.052 |
| 24 |  |
43 | 3.761 | 3.715 | 0.046 |
| 25 |  |
41 | 3.713 | 3.723 | −0.01 |
| 26 |  |
39 | 3.663 | 3.598 | 0.065 |
| 28 |  |
20 | 2.995 | 2.966 | 0.029 |
| 30 |  |
55 | 4.007 | 4.055 | −0.048 |
| 31 |  |
35 | 3.555 | 3.498 | 0.057 |
| 32 |  |
32 | 3.465 | 3.401 | 0.064 |
| 33 | 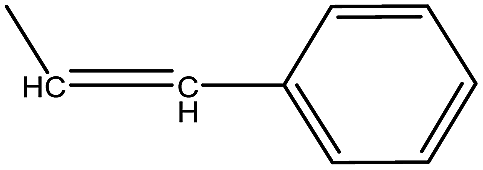 |
71 | 4.262 | 4.252 | 0.01 |
| 34 |  |
29 | 3.367 | 3.411 | −0.044 |
| 35 |  |
40 | 3.688 | 3.676 | 0.012 |
| 36 |  |
30 | 3.401 | 3.526 | −0.125 |
| 37 | 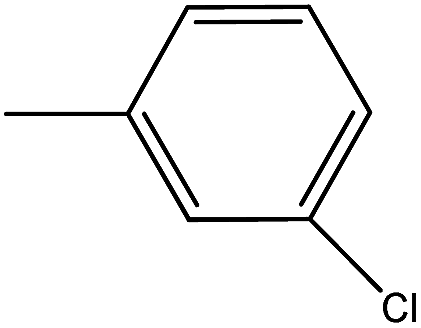 |
27 | 3.295 | 3.391 | −0.096 |
| 38 |  |
32 | 3.465 | 3.374 | 0.091 |
The structures of the synthesized compounds were confirmed by IR, mass and NMR analysis. The peak at about δ ppm 7.7–8.2 in 1H NMR spectra and the signal between 130 and 135 δ ppm in 13C NMR spectra confirm the CH at the fifth position of thiazolidine ring system. In principle two geometrical isomers, namely, E and Z are possible for all the Knoevenagel condensed products. However, the entire compounds exhibit only the Z configuration as expected from our previous studies. The reason for this deshielding is attributed to the cis position of the carbonyl function of highly electronegative rhodanine ring to the CH and hence the Z configuration. The cis positioning is due to the high degree of thermodynamic stability of these compounds because of the intramolecular hydrogen bond that can be formed between the hydrogen atom of CH and the oxygen atom in rhodanine31–33 (Scheme 1).
2.2. In vitro anticancer activity
Further, the compounds were subjected to in vitro anticancer screening against MCF-7 breast cancer cells using trypan blue dye exclusion test according to the standard protocol.34,35 The obtained % cytotoxicity values are when assessed at the concentration of 10 μg and are as shown in the Table 1. Among them, interestingly compounds 10, 22 and 33 with cinnamoyl substitution at the fifth position of rhodanine ring system showed significant cytotoxic activity against MCF-7 cells.2.3. CoMSIA study
Five-membered heterocyclic molecules containing thiazole nucleus with a carbonyl group on fourth carbon such as rhodanine derivatives have exhibited a broad spectrum of pharmacological activities. In this context, we subjected 33 N-substituted rhodanines to the 3D QSAR analysis for their anticancer activity against MCF 7 cells. The % cytotoxicity values were transformed to their natural logarithms (lnCyt) and used for the analysis. As part of the QSAR, we performed comparative similarity analysis CoMSIA as per the regular protocol using Sybyl-X 2.1.1. The molecules are analyzed for their conformations and ensured that all the compounds possess Z configuration. Added charges and aligned by atom fit method. The five similarity indices in CoMSIA, i.e., steric, electrostatic, hydrophobic, H-bond donor and H-bond acceptor descriptors were calculated and the fields generated. Partial least-squares (PLS)36 regression analyses were performed to generate the model. For the developed CoMSIA model, the cross-validated correlation coefficient (q2) value of the training set was 0.885 with six principal components. The non-cross-validated r2 value was 0.971 with a standard error of estimation (SEE) of 0.060 and a Fischer's covariance ratio (F) of 147.366 (significant at the 99% level). The predictive ability of the models was evaluated by leave-one-out (LOO) cross-validation. The developed model was further evaluated by predicting activities of the external test set compounds and the predictive r2 for the test set was 0.742, this indicates, good predictability of the developed model. Using the CoMSIA contour maps the structure–activity relationships are derived for N-substituted rhodanines for their anticancer activity. The developed 3D QSAR model is useful to predict the anticancer activity of newer rhodanines before their synthesis (Table 2).
| CPD No. | R1 | % cytotoxicity | lnCyt |
Predicted activity | Residual activity |
|---|---|---|---|---|---|
| 12 |  |
40 | 3.688 | 3.644 | 0.044 |
| 21 |  |
31 | 3.43 | 3.688 | −0.258 |
| 27 |  |
28 | 3.33 | 3.487 | −0.157 |
| 29 |  |
50 | 3.912 | 3.88 | 0.032 |
| 39 |  |
28 | 3.33 | 3.233 | 0.097 |
 | ||
| Fig. 2 Training set molecules after alignment by field fit method. | ||
 | ||
| Fig. 3 Correlation between the observed and predicted activities of the developed CoMSIA model. | ||
 | ||
| Fig. 4 CoMSIA steric SD × coefficient contour plot; green contours indicate regions where steric bulk is favorable and yellow contours indicate regions where steric bulk is not favored. | ||
The blue contour over the ortho and meta positions of the phenyl ring indicates that electronegative atoms like chlorine or oxygen will enhance the activity (Fig. 5). Compounds 8, 9, 11, 17, 18, 19, 20, 24, 25, 30, 31, 32, 35 and 36 with similar substitutions showed moderate to good activity. Interestingly, among the three series, compounds with orthochlorophenyl ring at the third position of the rhodanine showed better activity when compared to the cyclohexyl and benzyl substitutions.
 | ||
| Fig. 5 CoMSIA electrostatic SD × coefficient contour plot; blue contours indicate regions where electronegative groups increase activity and red contours indicate regions where electropositive groups increase activity (not contributed in the above contour). | ||
In Fig. 6, the yellow contour near the meta position of phenyl ring indicates that presence of hydrophobic halogen and methoxy group will enhance the activity (compounds 14, 17, 19 and 30). Similar substitution at ortho and para with hydrophobic groups has resulted in a reduction of activity (compound 8 and 9). Compounds with furan ring instead of phenyl ring system have failed to show the activity (compounds 16, 28 and 39). The white contour at the third and fifth position of the rhodanine ring indicates that increasing hydrophobicity in that region will not contribute to the activity. Specially, benzyl substitution at the third position of the rhodanine ring system has resulted in reduced activity. This is possibly due to the increased hydrophobicity in that region and steric extension. Evidence to this is, all the compounds of this series failed to show good activity (compounds 29 to 39).
 | ||
| Fig. 6 CoMSIA hydrophobic SD × coefficient contour plot; yellow contours indicate regions where hydrophobicity favors and white contours indicate regions where hydrophobicity disfavors. | ||
In Fig. 7, the large cyan contour (masked by violet contour) at the para position and part of meta position of the phenyl ring system at fifth position indicates that the presence of hydrogen bond donors will contribute to the activity (compounds 8, 20 and 31). Whereas, violet contour at other regions of the phenyl ring indicates disfavour for the hydrogen bond donors.
 | ||
| Fig. 7 CoMSIA hydrogen bond donor SD × coefficient contour plot; cyan contours indicate regions where hydrogen bond donor increase activity and violet contours indicate regions where hydrogen bond donor decrease activity. | ||
In Fig. 8, the large magenta contour near the meta position where nitro and oxygen present indicates that the hydrogen bond acceptors at that position are essential for the rhodanines to exhibit the activity. Similarly, the presence of magenta contour at the para position of phenyl ring also indicates hydrogen bond acceptors are partially favouring the activity (compounds 7, 17, 18, 19, 23, 29, 30 and 34).
 | ||
| Fig. 8 CoMSIA hydrogen bond acceptor SD × coefficient contour plot; magenta contours indicate regions where hydrogen bond acceptors increase activity and red contours indicate regions where hydrogen bond donors decrease activity (not contributed in the above contour). | ||
3. Conclusions
We have designed and synthesised a library of novel rhodanines for their anticancer activity. Rhodanines 10, 22 and 33 are considered to be the candidate compounds to investigate further, as they exhibited significant anticancer activity against MCF-7 human breast cancer cells. Present 3D QSAR model will be useful to design the new ligands of this class for their anticancer activity.4. Experimental
4.1. Chemistry
The synthesized compounds were characterized by MP, IR, NMR and MASS spectral analysis. TLC was performed using 2% ethyl acetate in chloroform as a mobile phase on aluminium plates precoated with silica gel GF. The melting points of the compounds were determined using melting apparatus by an open capillary method and are uncorrected. The IR spectra of the compounds were recorded on FT-IR spectrometer (Perkin Elmer Infrared-283) using KBr pellet technique. 1H NMR spectra ware recorded on AV-III (400 MHz FT-NMR) using DMSO-d6 as solvent and TMS as internal standard. The mass spectra were recorded using Shimadzu LCMS 2010A spectrometer under electrospray ionization technique. Microwave assisted synthesis was performed using scientific microwave system CATA R from Catalyst Systems (Pune, India).4.1.1.1. N-(2-Aminophenyl)(aminosulfanyl)carbothioamide (1). Pale yellow solid, yield 85%, mp 85–87 °C. FTIR (KBr, cm−1): 3324.84 (N–H), 3004.74 (Ar–H), 1664.12 (C
O, amide), 1384.64 (CS), 1019.45 (C–O). 1H NMR (δ ppm, CDCl3): 7.19–7.70 (m, 4H, ArH), 7.8 (s, 4H, NH4), 9.3 (s, 1H, NH). MS (m/z): M − 1 peak found 220.0 (M − 1 peak calculated 220.7). Mass fragments (m/z): 190.10, 211.00, 219.7.
4.1.1.2. N-Cyclohexyl(aminosulfanyl)carbothioamide (2). White amorphous solid, yield 75%, mp 173 °C. FTIR (KBr, cm−1): 3315.74 (N–H), 2854.74 (AliC–H), 1654.12 (C
O, amide), 1388.79 (CS), 1016.52 (C–O). 1H NMR (δ ppm, CDCl3): 1.15–1.52 (m, 10H, AliH), 3.12 (m, 1H, CH), 4.19–4.21 (d, 1H, NH). MS (m/z): M − 1 peak found 191.05 (M − 1 peak calculated, 192.35). Mass fragments (m/z): 182.10, 190.06, 191.06.
4.1.1.3. N-Benzyl(aminosulfanyl)carbothioamide (3). White crystalline solid, yield 90%, mp: 120–122 °C. FTIR (KBr, cm−1): 3061.13 (Ar C–H), 1371.43 (C–N), 1319.35 (C
S), 1683.91 (N–H). 1H NMR (δ ppm, CDCl3): 4.91 (s, 2H, ArCH2), 4.63–4.91 (m, 4H, NH4), 6.15 (s, 1H, NH), 7.26–7.42 (m, 5H, ArH). MS (m/z): M + 1 peak found 201.95 (M + 1 peak calculated 200.32). Mass fragments (m/z): 196.10, 198.03 and 190.10.
4.1.2.1. 3-(2-Aminophenyl)-2-thioxothiazolidin-4-one (4). White crystalline solid, yield 80%, mp 117–119 °C. FTIR (KBr, cm−1): 3091.99 (Ar C–H), 1732.13 (C
O, amide), 1477 (CS), 1386.86 (C–N), 1732.13 (CO), 698.25 (C–Cl). 1H NMR (δ ppm, CDCl3): 4.22 (d, 2H, CH2), 7.2–7.6 (m, 4H, ArH). MS (m/z): M − 1 peak found 241.20, (M − 1) peak calculated, 243.73. Mass fragments (m/z): 244.95, 243.96, 233.90, 220.95.
4.1.2.2. 3-Cyclohexyl-2-thioxothiazolidin-4-one (5). White amorphous solid, yield 85%, mp 70–72 °C. FTIR (KBr, cm−1): 2852.81 (AliC–H), 1691.63 (C
O, amide), 1602 (CC), 1396.51 (C–N), 1361.79 (CS), 1006.88 (C–O). 1H NMR (δ ppm, CDCl3): 1.202.19 (m, 11H, AliH), 4.1 (s, 2H, CH2), 4.38 (s, 1H, CH). MS (m/z): M − 1 peak found 215.95 (M − 1 peak calculated, 215.95). Mass fragments (m/z): 199.95, 181.05, 215.07.
4.1.2.3. 3-Benzyl-2-thioxothiazolidin-4-one (6). Yellowish amorphous solid, yield 89%, mp 70–75 °C. FTIR (KBr, cm−1): 1737.92 (C
O, amide), 1392.65 (C–N), 1319.35 (CS), 1033.88 (C–O). 1H NMR (δ ppm, CDCl3): 4.13 (s, 1H, CH2), 5.19 (s, 2H, CH2), 7.27–7.34 (m, 5H, ArH). MS (m/z): M + 1 peak found 225.05 (M + 1 peak calculated 223.31). Mass fragments (m/z): 219.00, 201.10, 190.10.
4.1.3.1. (Z)-3-(2-Chlorophenyl)-5-(4-methoxybenzylidene)-2-thioxothiazolidin-4-one (7). Yellow amorphous solid, yield 74%, mp 193–195 °C. FTIR (KBr, cm−1): 1718.63 (C
O, amide), 1635.69 (CC), 1477.52 (CS), 1361.79 (C–N), 1718.63 (CO), 1024.24 (C–O), 553.59 (C–Cl), 1361.79 (C–N). 1H NMR (δ ppm, CDCl3): 3.79 (s, 3H, OCH3), 7.02–7.70 (m, 8H, ArH), 7.80 (s, 1H, CH), 13C NMR: (δ ppm, CDCl3); 55.59, 123.05, 125.85, 127.11, 130.63, 130.55, 135.87, 114.21, 114.43, 127.41, 127.42, 127.57, 159.98, 142.15, 115.91, 166.89, 192.27.1H–1H-HOMO COSY: the signal at δ 7.02 was found to correlate with the off-diagonal signal at δ 7.51 and vice versa. This indicates that these two protons are neighboring aromatic protons. MS (m/z): M − 1 peak found, 359.90 (M − 1 peak calculated 361.87). Mass fragments (m/z):357.75, 338.80, 317.15.
4.1.3.2. (Z)-3-(2-Chlorophenyl)-5-(2-hydroxybenzylidene)-2-thioxothiazolidin-4-one (8). Yellow amorphous solid, yield 69%, mp 135–138 °C. FTIR (KBr, cm−1): 3078.49 (ArC–H), 1691.63 (C
O, amide), 1396.51 (C–N), 1356.00 (CS), 1047.38 (C–O), 576.74 (C–Cl). 1H NMR (δ ppm, CDCl3): 6.68–7.58 (m, 8H, ArH), 7.78 (s, 1H, CH), 9.1 (s, 1H, OH), 13C NMR (δ ppm, CDCl3): 123.21, 125.84, 129.13, 127.11, 130.52, 135.73, 115.82, 116.61, 121.31, 127.82, 129.41, 158.32, 141.43, 115.79, 166.88, 199.98. MS (m/z); M + 1 peak found, 347.97 (m + 1 peak calculated 348.14). Mass fragments (m/z): 346.95, 344.92, 340.95.
4.1.3.3. (Z)-5-(2-Chlorobenzylidene)-3-(2-chlorophenyl)-2-thioxothiazolidin-4-one (9). Yellow amorphous solid, yield 71%, mp 161–164 °C. FTIR (KBr, cm−1): 3053.42 (ArC–H), 1728.28 (C
O, amide), 1610.61 (CC), 1477 (CS), 1365.65 (C–N), 1047.38 (C–O), 565.15 (C–Cl). 1 H NMR (δ ppm, CDCl3): 7.20–7.70 (m, 8H, ArH), 7.78 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 123.21, 125.81, 127.13, 129.31, 130.72, 135.93, 126.82, 127.81, 128.81, 129.42, 131.21, 133.12, 142.43, 115.76, 166.81, 194.15. MS (m/z); M + 1 peak found, 365.97 (m + 1 peak calculated 366.14). Mass fragments (m/z): 364.95, 362.92, 358.95.
4.1.3.4. (Z)-3-(2-Chlorophenyl)-5-styryl-2-thioxothiazolidin-4-one (10). Yellow amorphous solid, yield 70%, mp 180–183 °C. FTIR (KBr, cm−1): 3061.13 (ArC–H), 1708.99 (C
O, amide), 1604.83 (CC), 1479.45 (CS), 1354.07 (C–N), 1047.38 (C–O), 586.38 (C–Cl). 1 H NMR (δ ppm, CDCl3): 6.72–6.85 (m, 2H, –CHCH–), 7.20–7.60 (m, 9H, ArH), 7.62 (d, 1H, CH), 13C NMR (δ ppm, CDCl3): 123.21, 125.87, 127.19, 129.51, 130.52, 135.73, 126.41, 126.81, 128.18, 128.32, 128.01, 135.12, 131.31, 125.3, 136.1, 119.12, 166.91, 193.35. MS (m/z); M + 1 peak found, 358.27 (m + 1 peak calculated 358.84). Mass fragments (m/z): 357.95, 354.92, 352.95.
4.1.3.5. (Z)-3-(2-Chlorophenyl)-5-(3-nitrobenzylidene)-2-thioxothiazolidin-4-one (11). Yellow amorphous solid, yield 80%, mp 260–263 °C. FTIR (KBr, cm−1): 3086.21 (Ar C–H), 1724.42 (C
O, amide), 1602.90 (CC), 1531.53 (NO2), 1477.52 (CS), 1350.22 (C–N), 1047.38 (C–O), 559.38 (C–Cl). 1H NMR (δ ppm, CDCl3): 7.30–8.4 (m, 8H, ArH), 7.77 (S, 1H, CH), 13C NMR (δ ppm, CDCl3): 120.32, 121.37, 129.49, 132.51, 148.32, 136.23, 142.13, 123.31, 125.81, 129.18, 130.32, 135.81, 127.12, 115.95, 166.91, 193.35. MS (m/z); M + 1 peak found, 376.97 (m + 1 peak calculated 377.14). Mass fragments (m/z): 375.95, 374.92, 373.95.
4.1.3.6. (Z)-3-(2-Chlorophenyl)-5-(2,4-dichlorobenzylidene)-2-thioxothiazolidin-4-one (12). Yellow amorphous solid, yield 82%, mp 193–195 °C. FTIR (KBr, cm −1): 3053.42 (ArC–H), 1726.35 (C
O, amide), 1604.83 (CC), 1479 (CS), 1388.79 (C–N), 1049 (C–O). 1 H NMR (δ ppm, CDCl3): 7.31–7.70 (m, 6H, ArH), 7.75 (s, 1H, CH), 8.1 (s, 1H, ArH), 13C NMR (δ ppm, CDCl3): 123.12, 125.87, 127.29, 129.21, 130.52, 135.83, 126.83, 127.83, 128.81, 129.48, 131.32, 133.11, 142.86, 115.95, 166.91, 193.35. MS (m/z); M + 1 peak found, 365.95 (m + 1 peak calculated 366.14). Mass fragments (m/z): 364.95, 366.92, 365.95.
4.1.3.7. (Z)-5-(4-Chlorobenzylidene)-3-(2-chlorophenyl)-2-thioxothiazolidin-4-one (13). Yellow amorphous solid, yield 80%, mp 200–222 °C. FTIR (KBr, cm−1): 3061.13 (ArC–H), 1712.85 (C
O, amide), 1602.90 (CC), 1357.93 (CS), 1404.22 (C–N), 1043.52 (C–O), 551.66 (C–Cl). 1H NMR (δ ppm, CDCl3): 7.30–7.80 (m, 8H, ArH), 7.85 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 123.02, 125.87, 127.29, 129.21, 130.52, 135.23, 127.83, 127.53, 128.8, 128.68, 133.32, 133.51, 142.86, 115.95, 166.91, 193.35. MS (m/z); M + 1 peak found, 365.95 (m + 1 peak calculated 366.14). Mass fragments (m/z): 364.95, 366.92, 365.95.
4.1.3.8. (Z)-5-(3-Chlorobenzylidene)-3-(2-chlorophenyl)-2-thioxothiazolidin-4-one (14). Yellow amorphous solid, yield 81%, mp 180–182 °C. FTIR (KBr, cm1): 1720.56 (C
O, amide), 1608.69 (CC), 1477.52 (CS), 1408.08 (C–N), 1720.56 (CO), 1031.95 (C–O), 557.45 (C–Cl). 1 H NMR (δ ppm, CDCl3): 7.25–7.7 (m, 8H, ArH), 7.79 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 123.22, 125.87, 127.19, 129.17, 130.41, 135.83, 124.43, 126.53, 128.12, 130.15, 134.23, 136.67, 142.96, 115.45, 166.96, 193.55. MS (m/z); M + 1 peak found, 365.91 (m + 1 peak calculated 367.14). Mass fragments (m/z): 364.85, 366.20, 362.90.
4.1.3.9. (Z)-5-Benzylidene-3-(2-chlorophenyl)-2-thioxothiazolidin-4-one (15). Yellow amorphous solid, yield 80%, mp 156–158 °C. FTIR (KBr, cm−1): 3080.42 (ArC–H), 1720.56 (C
O, amide), 1479.45 (CS), 1357.93 (C–N), 1031.95 (C–O), 553.59 (C–Cl). 1 H NMR (δ ppm, CDCl3): 7.2–7.7 (m, 9H, ArH), 7.85 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 123.12, 125.81, 127.12, 129.12, 130.54, 135.83, 126.43, 126.43, 128.82, 128.75, 128.73, 135.27, 142.76, 115.85, 166.92, 194.15. MS (m/z); M + 1 peak found, 332.18 (m + 1 peak calculated 331.84). Mass fragments (m/z): 316.85, 330.20, 326.90.
4.1.3.10. (Z)-3-(2-Chlorophenyl)-5-(furan-2-ylmethylene)-2-thioxothiazolidin-4-one (16). Yellow amorphous solid, yield 79%, mp 164–169 °C. FTIR (KBr, cm−1): 3051.4 (ArC–H), 1718.63 (C
O, amide), 1610.61 (CC), 1477.52 (CS), 1390.72 (C–N), 1024.24 (C–O), 601.811 (C–Cl). 1 HNMR (δ ppm, CDCl3): 6.65 (s, 1H, ArH), 6.90 (s, 1H, ArH), 7.29–7.68 (m, 5H, ArH), 7.80 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 123.00, 125.80, 127.10, 129.12, 130.51, 135.81, 111.41, 112.7, 145.93, 151.61, 142.61, 121.90, 166.9, 193.5. MS (m/z): M + 1 peak found 321.97 (M + 1 peak calculated, 322.19). Mass fragments (m/z): 320.85, 318.85, 322.90.
4.1.3.11. (Z)-3-(2-Chlorophenyl)-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxothiazolidin-4-one (17). Yellow amorphous solid, yield 68%, mp 191
193 °C. FTIR (KBr, cm−1): 1716.70 (CO, amide), 1610.61 (CC), 1431.23 (CS), 1356.17 (C–N), 1030.02 (C–O), 551.66 (C–Cl). 1H NMR (δ ppm, CDCl3): 3.73 (s, 3H, OCH3), 7.12–7.70 (m, 7H, ArH), 7.80 (s, 1H, CH), 9.0 (bs, 1H, OH), 13C NMR (δ ppm, CDCl3): 123.11, 125.81, 127.11, 129.11, 130.52, 135.82, 112.12, 116.83, 120.11, 128.82, 144.91, 151.32, 56.2, 142.43, 115.95, 166.78, 193.38. MS (m/z); M + 1 peak found, 377.97 (m + 1 peak calculated 378.14). Mass fragments (m/z): 376.95, 374.92, 372.95.
4.1.3.12. (Z)-3-Cyclohexyl-5-(4-methoxybenzylidene)-2-thioxothiazolidin-4-one (18). Yellow crystalline solid, yield 80%, mp 130–132 °C. FTIR (KBr, cm1): 2852.81 (AliC–H), 1703.20 (C
O, amide), 1381.08 (C–N), 1342.50 (CS), 1026.16 (C–O). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 3.85 (s, 3H, OCH3), 7.0 (d, 2H, ArH), 7.40 (d, 2H, ArH) 7.65 (s, H, CH). 13C NMR (CDCl3, δ): 25.08, 26.09, 24.96, 27.83, 27.52, 58.00, 114.89, 114.2, 127.23, 127.4, 127.61, 159.90, 55.9, 142.23, 115.51, 165.92, 192.18, MS (m/z): M + 1 peak found 334.80 (M + 1 peak calculated, 335.95). Mass fragments (m/z): 333.85, 334.85, 335.90.
4.1.3.13. (Z)-3-Cyclohexyl-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxothiazolidin-4-one (19). Pale yellow crystalline solid, yield 80%, and mp 130–132 °C. FTIR (KBr, cm−1): 2852.81 (AliC–H), 1703.20 (C
O, amide), 1342.50 (CS), 1386.86 (C–N), 1026.16 (C–O). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 3.90 (s, 3H, OCH3), 6.9 (s, 1H, ArH), 7.0 (d, 1H, ArH), 7.1 (d, 1H, ArH), 7.65 (s, 1H, CH), 9.1 (bs, 1H, OH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 112.00, 116.80, 120.12, 128.67, 144.43, 151.43, 56.3, 142.36, 115.97, 165.75, 192.19. MS (m/z): M + 1 peak found 350.80 (M + 1 peak calculated, 349.9). Mass fragments (m/z): 350.80, 348.90, 346.90.
4.1.3.14. (Z)-3-Cyclohexyl-5-(2-hydroxybenzylidene)-2-thioxothiazolidin-4-one (20). Yellow amorphous solid, yield 79%, mp 180–182 °C. FTIR (KBr, cm−1): 2856.67 (AliC–H), 1674.27 (C
O, amide), 1714.77 (CO), 1398.44 (C–N), 1332.86 (CS), 1004.95 (C–O). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 6.90 (d, 1H, ArH), 7.0 (t, 1H, ArH), 7.25 (t, 1H, ArH), 7.40 (d, 1H, ArH), 8.15 (s, 1H, CH), 9.0 (bs, 1H, OH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 115.8, 116.8, 121.8, 127.67, 129.43, 158.43, 142.36, 115.97, 165.75, 191.97. MS (m/z): M + 1 peak found 320.75 (M + 1 peak calculated 321.14). Mass fragments (m/z); 319.55, 320.45, 321.72.
4.1.3.15. (Z)-5-(2-Chlorobenzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one (21). Yellow amorphous solid, yield 79%, mp 180–182 °C. FTIR (KBr, cm−1): 2854.74 (AliC–H), 1708.99 (C
O, amide), 1386.86 (C–N), 1323.21 (CS), 1039.67 (C–O), 555.52 (C–Cl), 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 7.25 (d, 1H, ArH), 7.35 (d, 1H, ArH), 7.49 (t, 2H, ArH), 8.0 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 126.8, 127.8, 128.8, 129.67, 131.43, 133.43, 142.36, 115.97, 165.75, 192.87. MS (m/z): M + 1 peak found 338.75 (M + 1 peak calculated 339.17). Mass fragments (m/z); 337.55, 339.45, 338.72.
4.1.3.16. (Z)-3-Cyclohexyl-5-styryl-2-thioxothiazolidin-4-one (22). Yellow crystalline solid, yield 71%, mp 148–150 °C. IR (KBr, cm−1): 2854.74 (AliC–H), 1699.34 (C
O, amide), 1653.05 (CC), 1384.94 (C–N), 1319.35 (CS), 1024.24 (C–O). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 6.72–6.85 (m, 2H, –CHCH–), 7.10–7.50 (m, 5H, Ar H), 7.65 (d, 1H, CH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 126.4, 126.4, 128.00, 128.67, 128.7, 135.3, 131.2, 125.3, 136.43, 119.1, 165.95, 193.87. MS (m/z): M + 1 peak found 330.5 (M + 1 peak calculated 330.97). Mass fragments (m/z); 331.55, 329.45, 326.72.
4.1.3.17. (Z)-3-Cyclohexyl-5-(3-nitrobenzylidene)-2-thioxothiazolidin-4-one (23). Yellow amorphous solid, yield 70%, mp 238–240 °C. IR (KBr, cm−1): 2854.74 (AliC–H), 1708.99 (C
O, amide), 1602.90 (CC), 1537.32 (NO2), 1386.86 (C–N), 1334.78 (CS), 1041.60 (C–O). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 7.50 (s, 1H, ArH), 7.65 (d, 1H, ArH), 7.70 (t, 1H, ArH), 7.80 (d, 1H, ArH), 7.68 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 120.3, 121.4, 129.6, 132.67, 136.43, 148.43, 142.36, 119.67, 163.35, 193.87. MS (m/z): M + 1 peak found 349.75 (M + 1 peak calculated 349.97). Mass fragments (m/z); 345.55, 343.45, 340.72.
4.1.3.18. (Z)-3-Cyclohexyl-5-(2,4-dichlorobenzylidene)-2-thioxothiazolidin-4-one (24). Yellow amorphous solid, yield 80%, mp 138–140 °C. FTIR (KBr, cm−1): 2852.81 (AliC–H), 1716.70 (C
O, amide), 1626.05 (CC), 1386.86 (C–N), 1327.07 (CS), 1039.67 (C–O), 557.4 (C–Cl). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 7.25 (s, 1H, ArH), 7.35–7.40 (d, 2H, ArH), 7.90 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 126.9, 129.2, 130.21, 131.12, 132.54, 134.9, 143.36, 122.67, 163.15, 193.87. MS (m/z): M + 1 peak found 372.45 (M + 1 peak calculated 372.97). Mass fragments (m/z); 370.55, 368.45, 366.72.
4.1.3.19. (Z)-5-(4-Clorobenzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one (25). Yellow amorphous solid, yield 79%, mp 150–151 °C. FTIR (KBr, cm−1): 2854.74 (AliC–H), 1712.85 (C
O, amide), 1608.69 (CC), 1384.94 (C–N), 1323.21 (CS), 1010.73 (C–O), 592.17 (C–Cl). 1H NMR (δ ppm, CDCl3): 1.25–1.44 (m, 11H, AliH), 3.188 (s, 1H, CH), 7.26 (s, 1H, CH), 7.40–7.56 (m, 4H, ArH), 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 7.40–7.50 (d, 4H, ArH), 7.70 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 127.8, 127.8, 128.8, 128.8, 133.4, 133.5, 142.64, 121.87, 162.59, 193.17. MS (m/z): M + 1 peak found 338.45 (M + 1 peak calculated 338.87). Mass fragments (m/z); 336.55, 332.45, 330.72.
4.1.3.20. (Z)-5-(3-Chlorobenzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one (26). Yellow amorphous solid, yield 70%, mp 171–172 °C. FTIR (KBr, cm−1): 2854.74 (AliC–H), 1710.92 (C
O, amide), 1606.76 (CC), 1384.84 (C–N), 1321.28 (CS), 1001.09 (C–O), 669.32 (C–Cl). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 7.30 (s, 1H, ArH), 7.35–7.60 (m, 3H, ArH), 7.68 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 124.5, 126.45, 128.12, 130.12, 134.54, 136.75, 142.34, 121.90, 161.52, 193.17. MS (m/z): M + 1 peak found 338.25 (M + 1 peak calculated 338.97). Mass fragments (m/z); 336.85, 332.35, 330.82.
4.1.3.21. (Z)-5-Benzylidene-3-cyclohexyl-2-thioxothiazolidin-4-one (27). Yellow amorphous solid, yield 68%, mp 140–142 °C. FTIR (KBr, cm−1): 2854.74 (AliC–H), 1710.92 (C
O, amide), 1608.60 (CC), 1384.94 (C–N), 1330.93 (CS), 1039.67 (C–O). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 7.42–7.50 (m, 5H, ArH), 7.68 (s, 1H, CH), 1H NMR (δ ppm, CDCl3): 1.36–3.62 (m, 11H, CH2), 7.14–7.80 (m, 5H, Ar H), 7.26 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 126.20, 126.40, 128.17, 128.70, 128.70, 135.65, 142.34, 121.90, 161.52, 193.17. MS (m/z): M + 1 peak found 304.25 (M + 1 peak calculated 304.74). Mass fragments (m/z); 301.85, 396.85, 389.85.
4.1.3.22. (Z)-3-Cyclohexyl-5-(furan-2-ylmethylene)-2-thioxothiazolidin-4-one (28). Dark yellow amorphous solid, yield 81%, mp 181–182 °C. FTIR (KBr, cm1: 2856.67 (AliC–H), 1699.34 (C
O, amide), 1604.83 (CC), 1390.72 (C–N), 1319.35 (CS), 1026.16 (C–O). 1H NMR (δ ppm, CDCl3): 1.27–1.9 (m, 10H, CH2), 2.51 (bm, 1H, CH), 6.7 (d, 1H, ArH), 6.9 (d, 1H, ArH), 7.4 (t, 1H, ArH), 7.70 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 22.9, 22.9, 28.20, 30.4, 30.4, 56.6, 111.20, 112.32, 145.4, 151.42, 142.34, 121.90, 161.52, 193.17. MS (m/z): M + 1 peak found 293.05 (M + 1 peak calculated 294.7). Mass fragments (m/z); 291.85, 285.85, 282.85.
4.1.3.23. (Z)-3-Benzyl-5-(4-methoxybenzylidene)-2-thioxothiazolidin-4-one (29). Yellow amorphous solid, yield 80%, mp 142–145 °C. FTIR (KBr, cm−1): 3090.07 (Ar C–H), 1697.41 (C
O, amide), 1354.74 (C–N), 1336.71 (CS), 1030.02 (C–O). 1H NMR (δ ppm, CDCl3): 3.83 (s, 3H, OCH3), 5.29 (s, 2H, CH2), 7.0–7.55 (m, 9H, ArH), 7.70 (s, 1H, CH). 13C NMR (δ ppm, CDCl3): 47.48, 55.56, 114.97, 127.40, 128.07, 128.57, 128.95, 132.80, 133.44, 134.96, 119.89, 126.03, 161.78, 145.30, 167.98, 193.17. MS (m/z): M + 1 peak found 338.80 (M + 1 peak calculated 341.45). Mass fragments (m/z); 319.85, 303.85, 284.85.
4.1.3.24. (Z)-3-Benzyl-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxothiazolidin-4-one (30). Yellow amorphous solid, yield 79%, mp 151–154 °C. FTIR (KBr cm1): 3350.12 (O–H), 3061.13 (Ar C–H), 1705.13 (C
O, amide), 1375.29 (C–N), 1602.90 (CC), 1301.99 (CS), 1018.45 (C–O). 1H NMR (δ ppm, CDCl3): 3.90 (s, 3H, OCH3), 5.25 (s, 2H, CH2), 6.96–7.55 (m, 8H, ArH), 7.72 (s, 1H, CH), 9.1 (bs, 1H, OH), 13C NMR (δ ppm, CDCl3): 46.42, 56.2, 115.93, 127.00, 127.00, 126.80, 128.60, 128.60, 141.72, 113.5, 115.2, 120.28, 128.80, 145.6, 150.5, 142.56, 166.76, 192.79, MS (m/z): M + 1 peak found 358.12 (M + 1 peak calculated 358.86). Mass fragments (m/z): 358.12, 355.09, 353.97.
4.1.3.25. (Z)-3-Benzyl-5-(2-hydroxybenzylidene)-2-thioxothiazolidin-4-one (31). Yellow amorphous solid, yield 78%, mp 135–138 °C. FTIR (KBr, cm−1): 3070.78 (ArC–H), 1722.49 (C
O, amide), 1381.08 (C–N), 1321.28 (CS), 1031.95 (C–O). 1H NMR (δ ppm, CDCl3): 5.25 (s, 2H, CH2), 7.27–7.50 (m, 9H, ArH), 8.10 (s, 1H, CH), 9.02 (bs, 1H, OH), 13C NMR (δ ppm, CDCl3): 46.32, 115.93, 127.00, 127.00, 126.80, 128.60, 128.60, 141.72, 115.5, 116.7, 121.28, 127.80, 129.6, 158.5, 142.56, 166.76, 192.79, MS (m/z): M + 1 peak found 328.12 (M + 1 peak calculated 328.46). Mass fragments (m/z): 328.12, 326.09, 322.97.
4.1.3.26. (Z)-3-Benzyl-5-(2-chlorobenzylidene)-2-thioxothiazolidin-4-one (32). Yellow amorphous solid, yield 76%, mp 208–210 °C. FTIR (KBr, cm−1): 3084.28 (Ar C–H), 1680.05 (C
O, amide), 1344.43 (C–N), 1305.85 (CS), 1039.67 (C–O). 1H NMR (δ ppm, CDCl3): 5.28 (s, 2H, CH2), 7.20–7.60 (m, 9H, ArH), 8.0 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 46.12, 115.92, 127.00, 127.00, 126.80, 128.60, 128.60, 141.72, 126.28, 127.30, 128.6, 129.5, 131.1, 133.3, 142.12, 166.76, 192.79, MS (m/z): M + 1 peak found 346.12 (M + 1 peak calculated 346.46). Mass fragments (m/z): 345.12, 346.09, 343.97.
4.1.3.27. (Z)-3-Benzyl-5-styryl-2-thioxothiazolidin-4-one (33). Dark yellow amorphous solid, yield 75%, mp 170–175 °C. FTIR (KBr, cm−1): 3063.06 (ArC–H), 1708.99 (C
O), 1647.26 (CO, amide), 1375.29 (C–N), 1321 (CS), 1035.81 (C–O), 1321.28 (C–N), 1H NMR (δ ppm, CDCl3): 5.26 (s, 2H, CH2), 6.72–6.85 (m, 2H, –CHCH–), 7.06–7.42 (m, 10H, ArH), 7.78 (d, 1H, CH), 13C NMR (δ ppm, CDCl3): 46.12, 127.00, 127.00, 126.80, 128.60, 128.60, 141.72, 119.90, 136.1, 125.3, 131.2, 135.2, 126.4, 126.4, 128.00, 128.70, 166.7, 192.7, MS (m/z): M + 1 peak found 338.06 (M + 1 peak calculated 338.16). Mass fragments (m/z): 337.12, 338.09, 336.57.
4.1.3.28. (Z)-3-Benzyl-5-(3-nitrobenzylidene)-2-thioxothiazolidin-4-one (34). Pale yellow amorphous solid, yield 70%, mp 179–182 °C. FTIR (KBr, cm−1): 1708.99 (C
O), 1602.90 (CO, amide), 1375.29 (C–N), 1321.28 (CS), 1035.81 (C–O), 1H NMR (δ ppm, CDCl3): 5.30 (s, 2H, CH2), 7.10–7.90 (m, 9H, ArH), 7.80 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 46.12, 115.92, 127.00, 127.00, 126.80, 128.60, 128.60, 141.72, 120.30, 121.3, 129.6, 132.5, 136.1, 148.3, 142, 166.5, 192.7, MS (m/z): M + 1 peak found 357.12 (M + 1 peak calculated 356.96). Mass fragments (m/z): 357.12, 358.09, 356.97.
4.1.3.29. (Z)-3-Benzyl-5-(2,4-dichlorobenzylidene)-2-thioxothiazolidin-4-one (35). Bright yellow amorphous solid, yield 75%, mp 135–137 °C. FTIR (KBr, cm−1): 3086.21 (ArC–H), 1716.70 (C
O, amide), 1379.15 (C–N), 1300.07 (CS), 1039.67 (C–O), 613.38 (C–Cl), 1H NMR (δ ppm, CDCl3): 5.28 (s, 2H, CH2), 7.20–7.65 (m, 8H, ArH), 7.86 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 46.10, 115.92, 127.00, 127.00, 126.80, 128.60, 128.6, 141.70, 126.90, 129.2, 127.8, 130.3, 131.2, 132.6, 134.9, 142, 166.5, 192.7, MS (m/z): M + 1 peak found 380.12 (M + 1 peak calculated 379.86). Mass fragments (m/z): 380.96, 378.69, 379.97.
4.1.3.30. (Z)-3-Benzyl-5-(4-chlorobenzylidene)-2-thioxothiazolidin-4-one (36). Bright yellow amorphous solid, yield 71%, mp 155–157 °C. FTIR (KBr, cm−1): 3064.99 (ArC–H), 1708.99 (C
O, amide), 1373.36 (C–N), 1602 (CC), 1321.28 (CS), 1055.81 (C–O), 694.40 (C–Cl), 1H NMR (δ ppm, CDCl3): 5.29 (s, 2H, CH2), 7.22–7.64 (m, 9H, ArH), 7.78 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 46.09, 115.92, 127.00, 127.00, 126.80, 128.60, 141.70, 142.20, 127.8, 127.8, 128.8, 128.8, 133.3, 133.5, 166.5, 192.7, MS (m/z): M + 1 peak found 346.82 (M + 1 peak calculated 346.26). Mass fragments (m/z): 303.67, 344.19, 345.87.
4.1.3.31. (Z)-3-Benzyl-5-(3-chlorobenzylidene)-2-thioxothiazolidin-4-one (37). Yellow amorphous solid, yield 71%, mp 129–131 °C. FTIR (KBr, cm−1): 3061.13 (Ar C–H), 1712.85 (C
O, amide), 1379.15 (C–N), 1602.90 (CC), 1321.28 (CS), 1031.95 (C–O), 553.59 (C–Cl), 1H NMR (δ ppm, CDCl3): 5.28 (s, 2H, CH2), 7.15–7.60 (m, 9H, ArH), 7.76 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 47.02, 116.19, 124.5, 126.5, 128.10, 130.10, 134.20, 136.60, 127.00, 127.00, 126.80, 128.60, 128.60, 141.70, 166.9, 192.5, MS (m/z): M + 1 peak found 346.87 (M + 1 peak calculated 346.22). Mass fragments (m/z): 303.85, 343.10, 345.35.
4.1.3.32. (Z)-3-Benzyl-5-benzylidene-2-thioxothiazolidin-4-one (38). Yellow amorphous solid, yield 71%, mp 145–147 °C. FTIR (KBr, cm−1): 1710.92 (C
O, amide), 1600.97 (CC), 1384.94 (C–N), 1319.35 (CS), 1030.02 (C–O), 1H NMR (δ ppm, CDCl3): 5.28 (s, 2H, CH2), 7.08–7.66 (m, 10H, Ar H), 7.70 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 46.12, 115.9, 126.4, 126.4, 126.80, 127.00, 127.00, 128.00, 128.60, 128.60, 128.70, 135.20, 141.70, 166.7, 192.80, MS (m/z): M + 1 peak found 313.05 (M + 1 peak calculated 311.42). Mass fragments (m/z): 303.85, 283.10, 309.55.
4.1.3.33. (Z)-3-Benzyl-5-(furan-2-ylmethylene)-2-thioxothiazolidin-4-one (39). Pale yellow amorphous solid, yield 69%, mp 240–242 °C. FTIR (KBr, cm−1): 3040.47 (ArC–H), 1732.3 (C
O, amide), 1600.2 (CC), 1324.94 (C–N), 1321.4 (CS), 1030.7 (C–O), 1H NMR (δ ppm, CDCl3): 5.28 (s, 2H, CH2), 6.88–7.50 (m, 8H, ArH), 7.77 (s, 1H, CH), 13C NMR (δ ppm, CDCl3): 46.00, 111.4, 112.70, 121.90, 126.80, 127.00, 127.00, 128.60, 128.60, 141.70, 142.00, 145.9, 151.6, 166.5, 192.70, MS (m/z): M + 1 peak found 301.22, (M + 1 peak calculated 301.02). Mass fragments (m/z); 301.02, 298.43, 283.67.
4.2. In vitro anticancer activity
Anticancer activity of the compounds was evaluated by determining the percentage viability of MCF-7, human breast cancer cells using the trypan blue dye exclusion technique. MCF cells were cultured in the peritoneal cavity of healthy albino mice weighing 25 to 30 g by injecting a suspension of MCF cells (1 × 106 cells per ml), intraperitoneally. The cells were aspirated aseptically from the peritoneal cavity of the mice on day 15. The cells were washed with Hank's balanced salt solution (HBSS) and centrifuged for 10–15 min in the cooling centrifuge. The pellet was re-suspended with HBSS and the process was repeated three times. Finally, the cells were suspended in a known quantity of HBSS and the cell count was adjusted to 1 × 106 cells per ml. 0.1 ml of the diluted cell suspension was distributed into Eppendroff tubes and exposed to 0.1 ml of the test compound (10 μg) and incubated at 37 °C under 5% CO2 for 3 h. After 3 h, a trypan blue dye exclusion test was performed to determine the percentage viability. The pooled cells were mixed with 0.4% yield trypan blue in a ratio of 1:1 and the number of stained, non-stained and total number of cells were counted using a haemocytometer. Cell count taken from cells grown in the absence of the test compound was taken as 100% cell survival (control). Percentage cytotoxicity was calculated by using the formula for triplicate samples:| % cytotoxicity = (% viability of control − % viability of test)/(% viability of control) × 100. |
4.3. CoMSIA study
CoMSIA is a powerful and established tool for building 3D-QSAR models that can be applied to drug design.37 Three-dimensional structure building and all the modeling were carried out using the SYBYL-X 2.1.1 program package and the conformations of the compounds in the training and test sets were generated using the systematic conformational search method implemented in SYBYL-X 2.1.1. Energy minimization was affected using the Tripos force field38 with a distance-dependent dielectric and the Powell conjugate gradient algorithm with a convergence criterion of 0.001 kcal mol−1. Partial atomic charges were calculated by the Gasteiger–Huckel method.37,39 Consequently, all the N-substituted rhodanines were aligned according to their common substructure and using most active compound 10 as a template. Molecular alignment was affected with the field fit alignment40 method function of SYBYL. After consistently aligning the molecules within a lattice that extended 4 A° units beyond the aligned molecules in all directions with a grid step size of 2 A°, a probe sp3 carbon atom with a net charge of +1 and van der Waals radius of 1.52 A° was employed. The five similarity indices in CoMSIA, i.e., steric, electrostatic, hydrophobic, H-bond donor, and H-bond acceptor descriptors were calculated and the fields generated were scaled by the CoMSIA-STD method in SYBYL-X 2.1.1. Here, steric indices are related to the third power of the atomic radii, the electrostatic descriptors are derived from the atomic partial charges, the hydrophobic fields are derived from the atom-based parameters, and the H-bond donor and acceptor indices are obtained by a rule-based method based on the experimental results. In optimizing the CoMSIA performance, the most important parameter is how to combine the five fields in the CoMSIA model. To choose the optimal result, we systematically altered the combination of fields and chose the value that gave the best non-cross-validation, the smallest errors, and the largest F value. Finally, the model generated by combining the steric, electrostatic, hydrophobic, and hydrogen-bond acceptor and hydrogen bond donor fields was selected as the best CoMSIA model, and the contours were analyzed using this model. To derive the 3D-QSAR models, the CoMSIA descriptors were used as independent variables with the lnCyt activity value as a dependent variable. Partial least-squares (PLS)36 regression analysis was performed with the standard protocol implemented in the SYBYL package. The predictive ability of the models was evaluated by leave-one-out (LOO) cross-validation. The developed model was further evaluated by predicting activities of the external test set compounds.
References
- American Cancer Society and A. C. Society, J. Am. Chem. Soc., 2010, 8, 1–34 Search PubMed.
- Press Release, Int. Agency Res. Cancer, World Health Organization, 2013, pp. 12–14 Search PubMed.
- M. Bajaj, S. Suraamornkul, L. J. Hardies and L. Glass, Diabetologia, 2007, 50, 1723–1731 CrossRef CAS PubMed.
- R. Smith, V. Cokkinides and O. W. Brawley, Ca-Cancer J. Clin., 2009, 59, 27–41 CrossRef PubMed.
- A. K. Jain, A. Vaidya, V. Ravichandran, S. K. Kashaw and R. K. Agrawal, Bioorg. Med. Chem., 2012, 20, 3378–3395 CrossRef CAS PubMed.
- Y. Pandey, P. K. Sharma, N. Kumar and A. Singh, Int. J. PharmTech Res., 2011, 3, 980–985 CAS.
- D. Havrylyuk, L. Mosula, B. Zimenkovsky, O. Vasylenko, A. Gzella and R. Lesyk, Eur. J. Med. Chem., 2010, 45, 5012–5021 CrossRef CAS PubMed.
- W. Li, Y. Lu, Z. Wang, J. T. Dalton and D. D. Miller, Bioorg. Med. Chem. Lett., 2007, 17, 4113–4117 CrossRef CAS PubMed.
- S. Chandrappa, S. B. Benaka Prasad, K. Vinaya, C. S. Ananda Kumar, N. R. Thimmegowda and K. S. Rangappa, Invest. New Drugs, 2008, 26, 437–444 CrossRef CAS PubMed.
- C. Sawyers, Nature, 2004, 432, 294–297 CrossRef CAS PubMed.
- J. C. Magill, M. F. Byl, B. Goldwaser, M. P. Instructor, B. Yates, J. R. Morency, L. B. Kaban, W. C. Guralnick and M. J. T. Associate, J. Med. Devices, 2010, 3, 1–19 Search PubMed.
- C. Shiau, C. Yang, S. K. Kulp, K. Chen, C. Chen, J. Huang and C. Chen, Cancer Res., 2005, 65, 1561–1569 CrossRef CAS PubMed.
- Q. Li and W. Xu, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 53–63 CrossRef CAS PubMed.
- Y. Momose, T. Maekawa, T. Yamano, M. Kawada, H. Odaka and H. Ikeda, J. Med. Chem., 2002, 11, 1518–1534 CrossRef.
- L. F. Da Rocha Junior, M. J. B. De Melo Rêgo, M. B. Cavalcanti, M. C. Pereira, M. G. D. R. Pitta, P. S. S. De Oliveira, S. M. C. Gonçalves, A. L. B. P. Duarte, M. D. C. A. De Lima, I. D. R. Pitta and M. G. D. R. Pitta, BioMed Res. Int., 2013, 7, 1–8 Search PubMed.
- N. Swathi, Y. Ramu and K. Satyanarayana, Int. J. Pharm. Pharm. Sci., 2012, 4, 0–5 CAS.
- A. Chawla, H. Kaur, P. Chawla and U. S. Baghel, J. Global Trends Pharm. Sci., 2014, 5, 1641–1648 Search PubMed.
- O. Bozdaǧ-Dündar, Ö. Özgen, A. Menteşe, N. Altanlar, O. Atli, E. Kendi and R. Ertan, Bioorg. Med. Chem., 2007, 15, 6012–6017 CrossRef PubMed.
- A. Gupta, R. Singh, P. K. Sonar and S. K. Saraf, Biochem. Res. Int., 2016, 2016, 1–9 Search PubMed.
- S. G. Alegaon and K. R. Alagawadi, Med. Chem. Res., 2012, 21, 816–824 CrossRef CAS.
- M. Azizmohammadi, M. Khoobi, A. Ramazani, S. Emami, A. Zarrin, O. Firuzi, R. Miri and A. Shafiee, Eur. J. Med. Chem., 2013, 59, 15–22 CrossRef CAS PubMed.
- S. D. Knight, N. D. Adams, J. L. Burgess, A. M. Chaudhari, M. G. Darcy, C. A. Donatelli, J. I. Luengo, K. A. Newlander, C. A. Parrish, L. H. Ridgers, M. A. Sarpong, S. J. Schmidt, G. S. Van Aller, J. D. Carson, M. A. Diamond, P. A. Elkins, C. M. Gardiner, E. Garver, S. A. Gilbert, R. R. Gontarek, J. R. Jackson, K. L. Kershner, L. Luo, K. Raha, C. S. Sherk, C. M. Sung, D. Sutton, P. J. Tummino, R. J. Wegrzyn, K. R. Auger and D. Dhanak, ACS Med. Chem. Lett., 2010, 1, 39–43 CrossRef CAS PubMed.
- B. T. Moorthy, S. Ravi, M. Srivastava, K. K. Chiruvella, H. Hemlal, O. Joy and S. C. Raghavan, Bioorg. Med. Chem. Lett., 2010, 20, 6297–6301 CrossRef CAS PubMed.
- L. Xu, J. Vagner, J. Josan, R. M. Lynch, D. L. Morse, B. Baggett, H. Han, E. A Mash, V. J. Hruby and R. J. Gillies, Mol. Cancer Ther., 2009, 8, 2356–2365 CrossRef CAS PubMed.
- B. Meunier, Acc. Chem. Res., 2008, 41, 69–77 CrossRef CAS PubMed.
- K. Liu, W. Rao, H. Parikh, Q. Li, T. L. Guo, S. Grant, G. E. Kellogg and S. Zhang, Eur. J. Med. Chem., 2012, 47, 125–137 CrossRef CAS PubMed.
- A. G. Smith, K. A. Beaumont, D. J. Smit, A. E. Thurber, A. L. Cook, G. M. Boyle, P. G. Parsons, R. A. Sturm and G. E. O. Muscat, Int. J. Biochem. Cell Biol., 2009, 41, 844–852 CrossRef CAS PubMed.
- Y. Dai, World J Gastrointest Oncol, 2010, 2, 159 CrossRef PubMed.
- M. Tsujie, Exp. Cell Res., 2003, 289, 143–151 CrossRef CAS PubMed.
- V. Asati, D. K. Mahapatra and S. K. Bharti, Eur. J. Med. Chem., 2014, 87, 814–833 CrossRef CAS PubMed.
- B. R. Prashantha Kumar, N. Rashid Baig, S. Sudhir, K. Kar, M. Kiranmai, M. Pankaj and N. M. Joghee, Bioorg. Chem., 2012, 45, 12–28 CrossRef CAS PubMed.
- B. R. Prashantha Kumar and M. J. Nanjan, Bioorg. Med. Chem. Lett., 2010, 20, 1953–1956 CrossRef PubMed.
- B. R. Prashantha Kumar, M. D. Karvekar, L. Adhikary, M. J. Nanjan and B. Suresh, J. Heterocycl. Chem., 2006, 43, 897–903 CrossRef.
- S. Biswal, U. Sahoo, S. Sethy, H. K. S. Kumar and M. Banerjee, Asian J. Pharm. Clin. Res., 2012, 5, 1–6 CAS.
- K. Nathiya, S. S. Nath, J. Angayarkanni and M. Palaniswamy, Asian J. Pharm. Clin. Res., 2012, 5, 171–173 CAS.
- R. D. Cramer, J. D. Bunce, D. E. Patterson and I. E. Frank, Quant. Struct.-Act. Relat., 1988, 7, 18–25 CrossRef.
- K. C. Tsai, Y. C. Chen, N. W. Hsiao, C. L. Wang, C. L. Lin, Y. C. Lee, M. Li and B. Wang, Eur. J. Med. Chem., 2010, 45, 1544–1551 CrossRef CAS PubMed.
- M. Clark, R. D. Cramer and N. Van Opdenbosch, J. Comput. Chem., 1989, 10, 982–1012 CrossRef CAS.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- R. D. Cramer, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |