
Isolation and biomimetic total synthesis of tomentodiones A–B, terpenoid-conjugated phloroglucinols from the leaves of Rhodomyrtus tomentosa†
Abstract
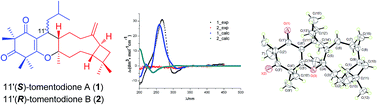
Tomentodiones A (1) and B (2), a pair of C-11′ epimers of caryophyllene-conjugated phloroglucinols with an unprecedented skeleton, were isolated from the leaves of Rhodomyrtus tomentosa. Their structures were elucidated through the application of extensive spectroscopic measurements with the absolute configuration of 1 determined by single-crystal X-ray diffraction analysis and electronic circular dichroism (ECD) calculations. The biogenetic pathways of 1 and 2 were proposed to involve an intermolecular, inverse electron demand Diels–Alder cycloaddition reaction as the key step, and their biomimetic total synthesis was accomplished.
 Please wait while we load your content...
Please wait while we load your content...

