
A facile access to novel heterocyclic analogues of chalcone from newly synthesized ketone containing isoxazole and a benzoxazinone ring†
 *a
*a
Abstract
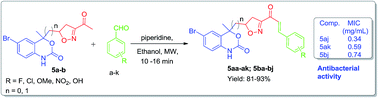
A series of novel heterocyclic analogs of chalcone containing isoxazole and a benzoxazinone ring as a prime motif was rationally designed and synthesized. There is nothing in the literature about such α,β-unsaturated ketones. These compounds were synthesized using a Claisen–Schmidt condensation reaction of the ketone precursor with different aromatic aldehydes using ethanol as solvent and piperidine as a base. Our procedure offers easy access to isoxazole and benzoxazinone based chalcone derivatives providing yields in the range of 81–93% within 16 min under mild conditions. Out of 21, 11 compounds were screened for their antibacterial activities. Among the screened compounds, compounds 5aj, 5ak and 5bj showed excellent antibacterial activities with MIC: 0.34, 0.59 and 0.74 mg ml−1 respectively as compared to standard drugs.
 Please wait while we load your content...
Please wait while we load your content...

