
Improved performance of a MnO2@PANI nanocomposite synthesized on 3D graphene as a binder free electrode for supercapacitors
Abstract
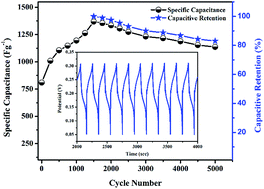
In the current study, we have synthesized a binder free nickel foam graphene MnO2@PANI (MnO2@PANI/graphene) nanocomposite by simple hydrothermal method followed by in situ polymerization. The graphene grown on nickel foam is used as a 3D scaffold conducting substrate for binder free nanocomposite electrode synthesis. Morphological characterization of the nanocomposite reveals growth of the MnO2 intercalated PANI nanorods-like network. The electrochemical study demonstrates very interesting electrode activation phenomenon, i.e. increase in specific capacitance upto 1500 charge–discharge cycles. The activated MnO2@PANI/graphene nanocomposite exhibits a maximum specific capacitance of 1369 F g−1 at 3 A g−1 current density and excellent rate capability of more than 70% by varying current density from 3 A g−1 to 15 A g−1. The activated electrode displays good cyclic stability by retaining SC over 83% after 5000 cycles.
 Please wait while we load your content...
Please wait while we load your content...

