
Screening and isolation of natural antioxidants from Ziziphora clinopodioides Lam. with high performance liquid chromatography coupled to a post-column Ce(iv) reduction capacity assay
Abstract
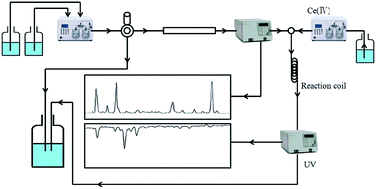
A novel on-line screening method for natural antioxidants was developed with a post-column cerium(IV) reduction reaction after high performance liquid chromatography (HPLC) separation. Under acidic conditions, Ce(IV) sulphate exhibits strong absorption at 320 nm and is reduced to Ce(III) by antioxidants in a post-column reaction coil (no absorbance at this wavelength). The reduction of absorbance at 320 nm affords a negative peak that corresponds to the retention time of an active constituent in the HPLC chromatogram. The proposed method demonstrated different selectivity versus a conventional HPLC-2,2-diphenyl-1-picrylhydrazyl (DPPH) method. This was used to detect the antioxidant constituents from the extract of Ziziphora clinopodioides Lam., which is an indigenous edible plant from north Xinjiang, China. Nine compounds were isolated as major antioxidant compounds by this method. All the isolated compounds were identified by 1H NMR and 13C NMR; six of them were isolated from this plant for the first time.
 Please wait while we load your content...
Please wait while we load your content...

