
A versatile in situ etching-growth strategy for synthesis of yolk–shell structured periodic mesoporous organosilica nanocomposites†
Yong Weia,
Xiaomin Lia,
Ahmed A. Elzatahryd,
Renyuan Zhangab,
Wenxing Wanga,
Xueting Tanga,
Jianping Yangac,
Jinxiu Wanga,
Daifallah Al-Dahyane and
Dongyuan Zhao*a
aDepartment of Chemistry and Laboratory of Advanced Materials, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Molecular Engineering of Polymers, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Shanghai 200433, P. R. China. Web: http://www.mesogroup.fudan.edu.cnE-mail: dyzhao@fudan.edu.cn; Fax: +86-21-51630307; Tel: +86-21-51630205
bSchool of Materials Science and Engineering, Key Laboratory of Advanced Civil Engineering Materials of Ministry of Education, Tongji University, 4800 Caoan Road, Shanghai, 201804, P. R. China
cState Key Laboratory of Pollution Control and Resources Reuse, College of Environmental Science and Engineering, Tongji University, Shanghai 200092, China
dMaterials Science and Technology Program, College of Arts and Sciences, Qatar University, P.O. Box 2713, Doha, Qatar
eDepartment of Chemistry-College of Science, King Saud University, Riyadh 11451, Saudi Arabia
First published on 17th May 2016
Abstract
This paper describes a versatile in situ etching-growth strategy for the preparation of periodic mesoporous organosilica (PMO) composites with yolk–shell structure, which can generate the void space and construct the outer PMO shells at the same time. The superparamagnetic yolk–shell Fe3O4@PMO composites (YS-Fe3O4@PMO) with radical mesochannels were also synthesized with this unique in situ etching-growth strategy by using Fe3O4@nSiO2 nanoparticles as the initial core. This method provides a general route for the synthesis of yolk–shell structured nanomaterials with different sized void spaces, various chemical composition cores, as well as organic functional PMO shells with radical mesochannels. Moreover, we can also obtain asymmetric or asymmetric hollow Fe3O4@PMO materials with a cubic PMO shell. All the magnetic mesoporous composites possess very high surface areas and large pore volumes (586 m2 g−1 and 0.52 cm3 g−1 for YS-Fe3O4@PMO, 946 m2 g−1 and 0.86 cm3 g−1 for asymmetric hollow Fe3O4@PMO). Gold nanoparticles could be encapsulated and confined in the void space of YS-Fe3O4@PMO composites through an in situ salt impregnation. The resultant YS-Fe3O4@Au@PMO nanomaterials could be used to catalyze the reduction of 4-nitrophenol with an ultrahigh efficiency (k = 0.01197 s−1). The magnetic catalysts could be easily recovered by a magnet and reused for more than 10 cycles with efficiency retained as high as 95%.
1. Introduction
Recently, nanomaterials with controlled and tailored properties1,2 have been considered to be powerful platforms for controlled release, confined nanocatalysis, energy storage and conversion.3–6 As one of the most important members in the nanomaterials family, magnetic nanocomposites7,8 with well-defined mesoporous structures, morphologies, and tailored properties have attracted immense scientific and technological interest, especially the nanocomposites with core–shell,9,10 yolk–shell11,12 and Janus13,14 structures. The syntheses of yolk–shell and Janus composites are usually more complicated, since their one-pot and direct synthesis is still limited. The yolk–shell nanoparticles or nano-rattles with core@void@shell structures,15–17 have attracted great scientific interest since the first report of this kind of structure,18 due not only to their tailorability and functionality in both the cores and shells, but also to the void space as nanoreactors and containers for drugs and medicine. Many routes and methods have been developed for the synthesis yolk–shell nanomaterials with a wide range of particle sizes, shapes, structures and chemical compositions.19–23 An important one is the selective etching approach from core–shell structured materials by dissolving or calcinating the inner shell of double-shelled nanocomposites,24,25 controlled dissolution/shrinkage of the core materials,26 structural difference-based selective etching strategy or surface-protected etching.27,28 Other routes such as ship-in-bottle approach,29 Ostwald ripening or galvanic replacement process,30 and Kirkendall effect31 based methods have also been proved as successful synthesis strategies for the yolk–shell structured materials.The periodic mesoporous organosilica (PMO) materials32,33 with organic moieties uniformly distributed in the whole frameworks, have attracted widespread interests due to their well-designed hydrophobicity and hydrophilicity,34 as well as the excellent biocompatibility and biodegradation.35,36 And thus the PMO materials have been proposed as promised materials for heterogeneous catalysis,37 drug delivery systems,38 chromatography,39 and enzyme immobilization,40 etc. Although there have been several routes for one-step synthesis of the yolk–shell structured composites with silica shells,41–43 many of these methods have resulted in unexpected nonporous silica shells or mesoporous silica without ordered mesopore channels. Some routes have been reported for the synthesis of PMO nanocomposites with core–shell and yolk–shell nanostructures such as the sol–gel process for the growth of PMO shell on up-conversion nanoparticles (UCNPs),44 dense silica spheres,45,46 cobalt and iron oxide nanoparticles,42,47–49 as well as the hydrothermal treatment assisted transformation from core–shell structured PMO materials45 and organosilane replacement46,50 to generate yolk–shell PMO composites. However, several problems are still needed to be settled with, one is that the mesoporous shells always have disordered mesostructures, and another is difficult to obtain different nanostructures (core–shell, yolk–shell and asymmetric nanostructure) from the same system. So new routes for the facile and controllable synthesis of mesoporous organosilica composites with tailored functionality and controlled structure are urgently desired.
Herein, we report a facile synthesis of ordered mesoporous organosilica composites with a yolk–shell structure and controllable PMO shell through an in situ etching-growth strategy. Uniform yolk–shell structured mesoporous composites with movable cores of different compositions and morphologies, void space of different sizes, and ethane- or benzene-bridged PMO shells can be obtained by using this strategy. Moreover, the asymmetric Fe3O4@PMO materials can also be obtained via slightly tuning the alkalinity of the solution. The uniform superparamagnetic yolk–shell Fe3O4@PMO composites (YS-Fe3O4@PMO) have radical mesopore nanochannels, while the asymmetric hollow Fe3O4@PMO composites (AH-Fe3O4@PMO) have crystal-like cubic PMO components (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) n symmetry). All the materials possess high surface areas (586–946 m2 g−1), large pore volumes (0.52–0.86 cm3 g−1) and are magnetic separable (7.0–11.5 emu g−1). Furthermore, gold nanoparticles could be encapsulated and confined in the void space of the yolk–shell PMO composites through a salt impregnation method and the resulted YS-Fe3O4@Au@PMO nanomaterials could be used to catalyze the reduction of 4-nitrolphenol with an ultrahigh efficiency and recyclability.
n symmetry). All the materials possess high surface areas (586–946 m2 g−1), large pore volumes (0.52–0.86 cm3 g−1) and are magnetic separable (7.0–11.5 emu g−1). Furthermore, gold nanoparticles could be encapsulated and confined in the void space of the yolk–shell PMO composites through a salt impregnation method and the resulted YS-Fe3O4@Au@PMO nanomaterials could be used to catalyze the reduction of 4-nitrolphenol with an ultrahigh efficiency and recyclability.
2. Experimental section
2.1 Chemicals
Gold chloride (HAuCl4·3H2O), bis-(triethanoxylsilyl)benzene (BTEB, 96%) and silver nitrate (AgNO3) were purchased from Aldrich and bis-(triethanoxylsilyl)ethane (BTSE, 96%) from Meryer. All other reagents such as concentrated ammonia (NH4OH, 28 wt%), hexadecyltrimethylammonium bromide (CTAB), tetraethanoxylsilane (TEOS, >99%) were purchased from Guoyao Chemical Company. All reagents were used as received without any further purification.2.2 Synthesis
Dense silica spheres (DS, ∼160 nm), silver nanocubes coated with a silica shell (Ag@nSiO2) and Fe3O4@nSiO2 nanoparticles (∼120 nm) with smaller Fe3O4 cores (∼60 nm in diameter) were used to synthesize the PMO composites with yolk–shell structure following the similar procedure above. The resultant YS-A@PMO composites (A reveals the core material), i.e., YS-60Fe3O4@PMO, YS-DS@PMO and YS-Ag@PMO were further extracted and measured with several methods.
2.3 Catalytic reduction of 4-nitrophenol
The catalytic reaction was carried out as follows. 20 mg of catalysts was dispersed in a quartz cuvette containing 35 mg portion of 4-nitrophenol (4-NP) solution (0.0757 g of 4-NP dissolved in 100 mL of H2O) and 1.00 g of NaBH4 solution (0.02 g in 3.0 mL of H2O) with total volume of 2 mL for the solution, and a UV-vis spectroscopy was used to monitor the absorption changes during the reaction. The particles were later recovered to test the cyclic catalytic performance.2.4 Characterization
Powder X-ray diffractions (XRD) measurements were performed on a Bruker D8 diffractometer using Cu Kα radiation at 30 kV and 15 mA. High-resolution scanning electron microscopy (HRSEM) images and energy dispersive X-ray analysis (EDX) were recorded on a Philips S4800 microscope operated at 10 kV, and transmission electron microscopy (TEM) images were obtained by JEOL JEM 2010 electron microscope with an acceleration voltage 200 kV. The powder samples for the TEM measurements were suspended in ethanol by ultrasonication and then dropped onto the Cu grids with holey carbon films. Nitrogen sorption isotherms for samples were obtained with a Micromeritics ASAP 2420 analyzer at 77 K after out-gassed at 120 °C for 10 h. The Barrett–Emmett–Teller (BET) specific surface areas were calculated using adsorption data in a relative pressure range of P/P0 between 0.05 and 0.25. Pore size distributions were derived from the adsorption branch using BJH method. The total pore volumes were estimated from the amounts adsorbed at highest relative pressure (P/P0 > 0.98). X-ray photoelectron spectroscopy (XPS) experiments were carried out on a RBD upgraded PHI-5000C ESCA system (Perkin Elmer) with Mg Kα radiation (hν = 1253.6 eV). The samples were directly pressed to a self-supported disk (10 × 10 mm) and mounted on a sample holder then transferred into the analyzer chamber. The whole spectra (0–1200 eV) and the narrow spectra of all the elements with much high resolution were both recorded by using RBD 147 interface (RBD Enterprises, USA) through the AugerScan 3.21 software. Binding energies were calibrated by using the containment carbon (C1s = 284.6 eV). The data analysis was carried out by using the XPSPeak 4.1. Solid-state 29Si MAS NMR experiments (79.4 MHz) were recorded on a Bruker MSL-300 spectrometer. The spectrometer was equipped with a 4 mm double air bearing, magic-angle spinning probe for MAS experiments. 29Si MAS NMR experiments were performed with a contact time of 5 ms, spin rate of 8 kHz, repetition delay of 3 s and Q8M8 ([(CH3)3SiO]8Si8O12) as a reference sample. Magnetic properties were determined using a SQUID VSM (superconducting quantum interference device, MPMS-VSM, from the Quantum Design Company, USA) magnetometer.3. Results and discussion
In the synthesis of the yolk–shell structured mesoporous composites with PMO shells, the Fe3O4 nanoparticles were selected as the representative core because of its magnetic feature as well as the high contrast between the nanoparticles and the PMO shells, which makes it convenient for not only the separation and recovery but also the TEM observations. The synthesis procedure of the yolk–shell structured YS-Fe3O4@PMO mesoporous composites with a magnetic Fe3O4 core, radical mesochannel PMO shell and controlled void space is represented in Scheme 1. Firstly, the Fe3O4 nanospheres were synthesized via a solvothermal method and then coated with a thin layer of non-porous silica shells to form core–shell structured Fe3O4@nSiO2 nanocomposites by the Stöber method (Scheme 1a). Later, the Fe3O4@nSiO2 nanospheres were used to synthesize the Fe3O4@PMO nanocomposites (Scheme 1b and c) in an aqueous solution of cationic surfactant CTAB with BTSE as a silica precursor and NaOH as the alkaline source. The hydrated organosilane oligomers co-assembled with the cationic surfactant micelles to direct the perpendicular growth of the PMO shells, and the non-porous silica shells of the cores were gradually etched at the same time (Scheme 1b). During the process, the condense silica shells were totally etched, and partial of the dissolved silica species were also introduced to co-construct the PMO shells which became thicker and thicker, until the separation of the magnetic mesoporous PMO composites (Scheme 1c). At the end, the Fe3O4@PMO composites were extracted with ethanol for further characterizations (Scheme 1d). | ||
| Scheme 1 Schematic illustration of the mechanism for the formation of yolk–shell Fe3O4@PMO nanocomposite via the “in situ etching-growth” strategy. (a) Coating non-porous silica shells (nSiO2) on the magnetic Fe3O4 cores (the Stöber method); (b) primary growth of PMO shells via surfactant/organosilane micelle self-assembly as well as partially etching of the nonporous silica shells at the solid–liquid interface; (c) further growth of PMO shells and totally etching of the nonporous silica shells; (d) extraction of the mesostructural template CTAB by ethanol at 60 °C. | ||
The Fe3O4 nanospheres (Fig. S1†) prepared by the solvothermal method51 have uniform particle sizes of ∼150 nm. After coating a thin-layer condense silica, the obtained core–shell structured Fe3O4@nSiO2 nanocomposites are also very uniform with a diameter of ∼200 nm and a nonporous silica layer of ∼25 nm in thickness as shown in the SEM images (Fig. 1a and b). After further PMO coating process, the Fe3O4@PMO samples also exhibit mono-dispersed and discrete particles with a spherical morphology (Fig. 1c). Its hollow interior can be clearly observed from a broken particle (inset Fig. 1c), indicating that the hybrid composites present yolk–shell structure with outer PMO shells and movable magnetic Fe3O4 cores (YS-Fe3O4@PMO). The yolk–shell nanoparticles have an overall diameter of ∼320 nm (Fig. 1c–e). The outer PMO layers with ordered radical mesopore channels exhibit a uniform shell thickness of ∼60 nm (Fig. 1d and e), the mesopore size of the PMO shells is evaluated to be 2–3 nm, which is in agreement with the size of CTAB micelles. The amorphous condense silica shells of the initial Fe3O4@nSiO2 nanoparticles disappear after the coating process of PMO shells and a hollow interior with a size of ∼50 nm appears. In addition, the inner diameter of the PMO shells is close to the size of the primary Fe3O4@nSiO2 nanoparticles (∼200 nm) and the size of the void space is estimated to be twice of the thickness for the nonporous silica shells (Fig. 1b and e).
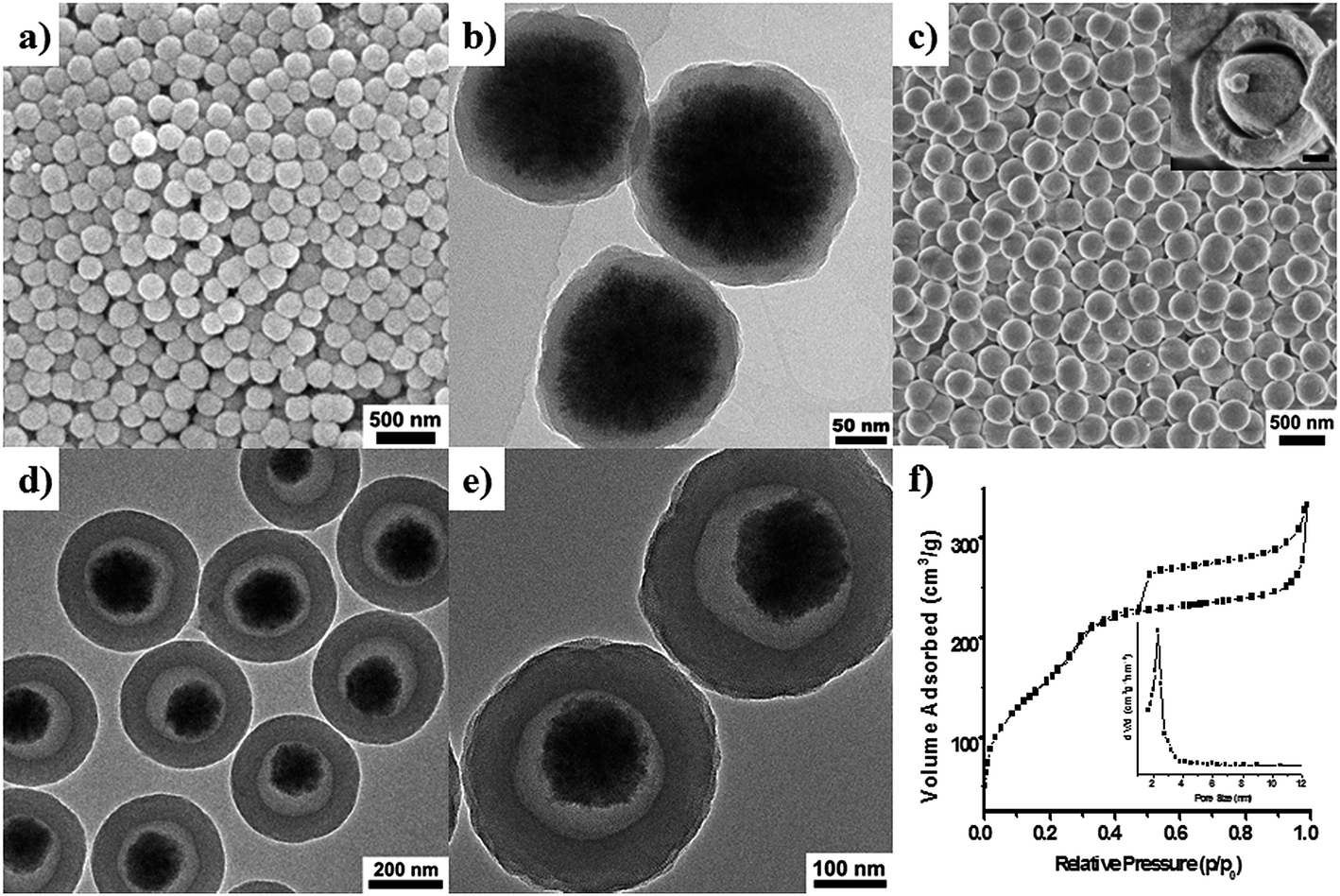 | ||
| Fig. 1 Characterization of the core–shell Fe3O4@nSiO2 nanoparticles and typical yolk–shell YS-Fe3O4@PMO mesoporous composites. SEM (a) and TEM (b) images of the core–shell Fe3O4@nSiO2 nanoparticles; HRSEM (c), TEM (d and e) images, nitrogen adsorption–desorption isotherms (f) and corresponding pore size distributions (inset (f)) of the yolk–shell YS-Fe3O4@PMO composites with Fe3O4 nanoparticles (∼150 nm in diameter) as the core. The HRSEM image inset (c), showing a broken particle of the YS-Fe3O4@PMO (the scale bar is 50 nm). | ||
The small-angle XRD patterns (SAXRD) (Fig. S2a†) of the yolk–shell YS-Fe3O4@PMO products present a broad peak at 2θ = 2.21°, suggesting a low-regular mesostructure of the PMO shells, in consist with the radical mesopore channels in the PMO shells. The nitrogen adsorption–desorption isotherms (Fig. 1f) of the typical YS-Fe3O4@PMO materials show type IV adsorption feature, revealing the abundant mesopores in the PMO shells. The BET specific surface area and total pore volume of the YS-Fe3O4@PMO composites are calculated to be as high as 586 m2 g−1 and 0.52 cm3 g−1, respectively, which can be attributed to the lower mass density of ethane silica-based frameworks, the well-arranged mesochannels and the inner void space. The mesopore size is centered at ∼2.2 nm based on BJH model from the adsorption branch (Fig. 1f, inset). In addition, its H2/H4 hybrid type hysteresis indicates the formation of a hollow structure with mesopore walls,54 further revealing the void space of the yolk–shell YS-Fe3O4@PMO nanocomposites, in accord with the HRSEM and TEM results. The wide-angle XRD patterns (WAXRD) (Fig. S2b†) of the yolk–shell YS-Fe3O4@PMO composites show one broad diffraction peak at 2θ near to 21° and six sharp peaks at 2θ = 30.28, 35.30, 43.37, 53.69, 57.27, 62.78°, corresponding to the reflections of amorphous silica and [220], [311], [400], [422], [511], [440] diffractions of Fe3O4 phase, respectively. The products retain their superparamagnetic property after the coating and extraction processes with saturated magnetization of ∼11.5 emu g−1 (Fig. S3†), which makes it convenient for further magnetic collection and also its recovery after applications (Fig. S4†). The 29Si MAS-NMR spectrum (Fig. S5†) of the yolk–shell YS-Fe3O4@PMO sample after the etching of the magnetic cores by HCl, shows two sharp bands at around −55.7 and −66.4 ppm, which can be contributed to T2 and T3 species of the ethane-bridged frameworks. The two weak bands between −120 and −100 ppm can be assigned as Q3 and Q4 sites of pure silica. Although Q3 and Q4 signals are not so apparent because the amount of pure silica is very low relative to the organosilica (Q/T < 5% based on the peak areas), clearly indicating the partial incorporation of the etched silica species into the final PMO shells. It further suggests that the silica layer of the core materials is etched during the synthesis process, and later-on co-assembled with the organosilane species and surfactants in the outer PMO shells.
As supposed before, the hollow interior of the yolk–shell mesoporous composites derived from the outer silica shells of the core materials, and the void space could be tuned easily by varying the former silica shell thickness. So that Fe3O4@nSiO2 nanospheres with different shell thickness can be used as the cores to construct yolk–shell magnetic nanocomposites. When the thickness of amorphous silica shells is ∼10 nm (Fig. 2a), the size of the void space in the final composites can be ∼20 nm (Fig. 2c); and the hollow interior is ∼80 nm (Fig. 2d) in size when the condense silica shell has a thickness of ∼40 nm (Fig. 2b). The hollow space is seemly twice of the shell thickness which is well in accord with that we proposed early. On the other side, the void space of the magnetic composite can be also tuned by the time interval between the injection of NaOH and BTSE, and the resultant composites were labeled as YS-Fe3O4@PMO-t, where t reveals the time interval in minute. At an interval of 30 s (0.5 min), the resultant yolk–shell Fe3O4@PMO composites (YS-Fe3O4@PMO-0.5) have a void space with relatively smaller size of ∼35 nm and a PMO shell with a thickness of ∼45 nm (Fig. 3a–c). As the interval increases to 2 min, the YS-Fe3O4@PMO-2 composites have even smaller void space of approximately 15 nm (Fig. 3d–f), while the YS-Fe3O4@PMO-5 composite shows a core–shell structure with no hollow interior and a PMO shell of ∼50 nm (Fig. 3g–i). These results indicate that in the present synthesis system, the amorphous condense silica shell (∼25 nm) of the Fe3O4@nSiO2 cores could be totally etched within 5 min.
 | ||
| Fig. 2 Characterization of the core–shell Fe3O4@nSiO2 nanoparticles and the corresponding yolk–shell YS-Fe3O4@PMO spheres with different void spaces. TEM images of the core–shell Fe3O4@nSiO2 nanoparticles with controlled silica shell thickness: (a) ∼10 nm, (b) ∼40 nm; TEM images of the yolk–shell YS-Fe3O4@PMO nanoparticles with different void spaces: (c) ∼20 nm, (d) ∼80 nm. | ||
 | ||
| Fig. 3 Characterization of the magnetic PMO composites YS-Fe3O4@PMO-t with different time interval between the addition of NaOH and BTSE (t = 0.5, 2, 5). SEM and TEM images of the yolk–shell YS-Fe3O4@PMO-0.5 (a–c), YS-Fe3O4@PMO-2 (d–f), YS-Fe3O4@PMO-5 (g–i) composites. The insets show the structural models of the yolk–shell YS-Fe3O4@PMO composites with different void space. | ||
Based on the above results, we propose an “in situ etching-growth” strategy for the formation of PMO composites with uniform yolk–shell structure. Firstly, the cationic surfactant micelles can be adsorbed onto the outer surfaces of the amorphous silica shells by electronic interaction. After NaOH and BTSE are injected into the aqueous solution containing CTAB surfactant and Fe3O4@nSiO2 nanospheres, the hydrated organosilane oligomers and the adsorbed surfactants micelles can assemble at the liquid–solid interface between the cores and solvents, and form mesostructured PMO shells. At the same time, the outer dense silica shells of the Fe3O4 cores are gradually etched under high alkaline conditions (pH ≈ 12) at a high temperature (80 °C). The silica layer is gradually dissolved and become thinner, according to the results with different time interval, the silica shell with the thickness of ∼25 nm can totally be etched in less than 5 minutes. The formed silica species can diffuse out, co-assemble with the organosilane/CTAB micelles, to co-construct the outer PMO shell and make it thicker and thicker until the silica shell is totally etched. Finally, all the former silica shells disappear and the ordered PMO shells successfully grow, and uniform yolk–shell nanocomposites with a void space between the cores and the shells are obtained. When the organosilane is added some time later after the addition of NaOH (such as 0.5 or 2 min), the Fe3O4@nSiO2 cores can be partially etched, the organosilane-surfactant micelles can be assembled at the freshly formed surfaces of the partially etched cores, and PMO shells further grow as well as the etching of the residual amorphous silica layer, therefore the yolk–shell nanocomposites with smaller void spaces are obtained. When the organosilane is added at certain time as the silica layer of the cores are totally etched, the surfactant micelles can be assembled at the external surface of the Fe3O4 nanoparticles, and the core–shell structured mesoporous composites are obtained. In this process, the nonporous silica shells serve as the sacrificial layer for the formation of the hollow interior. The supposed strategy helps to understand the precise control of the void space via tuning the thickness of the silica shells and the change of the void space at different time interval.
The “in situ etching-growth” strategy can be extended to synthesize yolk–shell mesoporous organosilica composites by using core materials with different sizes, chemical compositions and shapes. Dense silica spheres (dSiO2), silica coated silver nanocubes (Ag@nSiO2) and Fe3O4@nSiO2 nanospheres with smaller Fe3O4 cores can be employed as the cores for further growth of PMO shells (Fig. S6†). The yolk–shell PMO composites with dense silica sphere as the core (YS-DS@PMO) are synthesized from dense silica spheres with the size of ∼160 nm (Fig. S7a†), the resultant YS-DS@PMO particles have ∼260 nm in size (Fig. S6a†). The YS-DS@PMO sample has a void space of ∼40 nm and the outer shell with radical ordered mesopore channels (Fig. S6b†). When Ag@nSiO2 nanoparticles (Fig. S8†) with silver nanocubes of ∼50 nm and silica shells of ∼5 nm are used as the initial cores, the obtained YS-Ag@PMO particles have a diameter of ∼120 nm (Fig. S6c†), and a small void space of ∼10 nm (Fig. S6d†). The yolk–shell YS-60Fe3O4@PMO magnetic mesoporous composites with Fe3O4 cores of ∼60 nm and void space of ∼30 nm (Fig. S6e and f), can be obtained by using the Fe3O4@nSiO2 nanoparticles (Fig. S8†) with the size of 120 nm (Fe3O4 core of ∼60 nm and silica shell of ∼30 nm) as the core sources. The nitrogen isotherms (Fig. S9†) of the YS-DS@PMO, YS-Ag@PMO and YS-60Fe3O4@PMO materials also show high specific BET surface areas (400–600 m2 g−1), large pore volumes (0.4–0.5 cm3 g−1) and similar mesopore sizes (∼2.2 nm). Moreover, the PMO shells can also be constructed by other bridged organosilanes. Magnetic mesoporous composites with yolk–shell structure can be also obtained as benzene-bridged organosilane (BTEB) is used instead of BTSE. The uniform yolk–shell structure can clearly be observed from SEM images (Fig. S10a†) because of the high contrast among the core, the hollow spaces and the PMO shells are further confirmed by TEM images (Fig. S11†). However, when TEOS is used as a silica source to build the outer silica shells, only silica rods with a large size of ∼1 μm are obtained (Fig. S10b†), indicating that only bridged organosilanes are suitable to build the yolk–shell nanostructures under the present synthesis condition.
At similar conditions, while the amount of NaOH is reduced (e.g. 0.25 mL), the anisotropic growth of the PMO component lead to magnetic composite with PMO truncated-cubes (Fig. 4a) partially coated on Fe3O4 cores, and thus the resulted composite is asymmetric. The Fe3O4@PMO crystals with asymmetric hollow structure (AH-Fe3O4@PMO) are highly mono-dispersed and uniform with a narrow size distribution (∼600 nm), they also possess unique crystal-like external shape with the cores, which can be clearly seen from HRSEM images (Fig. 4b). The crystals have a hollow space with the same size (∼50 nm) as typical YS-Fe3O4@PMO materials (Fig. 4c), and the PMO component have ordered mesostructure (Fig. S12†) with cubic Pmn symmetry (Fig. S13†). The AH-Fe3O4@PMO composites have a surface area of ∼946 m2 g−1, pore volume of ∼0.86 cm3 g−1, and bimodal mesopore size distributions centered at 4.1/5.6 nm (Fig. S14†). The asymmetric AH-Fe3O4@PMO composites are also retained the super-paramagnetic property with the saturated magnetization of ∼7.0 emu g−1 (Fig. S3†). Similarly, the asymmetric Fe3O4@PMO composites without a void space (AS-Fe3O4@PMO) can be synthesized while much more Fe3O4@nSiO2 nanoparticles (4 times) are used to grow the magnetic PMO mesoporous composites with cubic mesoporous shells (Fig. 4a, process II). The AS-Fe3O4@PMO nanoparticles also have discrete and uniform crystals with the size of ∼400 nm (Fig. 4d and e) and ordered PMO components with cubic mesostructure (Fig. 4f) partially coated on the cores. The AS-Fe3O4@PMO products have discrete particles of ∼400 nm (Fig. 4d and e), and the PMO components also exhibit cubic mesostructure (Fig. 4f). The crystals show no hollow interior (Fig. 4d–f) mainly attributed to the excess silica species of the primary silica shells.
 | ||
| Fig. 4 Illustrative scheme and characterization for the synthesis of the magnetic asymmetric Fe3O4@PMO nanocomposites with cubic PMO shells. Schematic illustration (a) for the synthesis of the asymmetric Fe3O4@PMO crystals with a hollow interior (AH-Fe3O4@PMO) and without a void space (AS-Fe3O4@PMO), SEM and TEM images of the AH-Fe3O4@PMO (b and c) and AS-Fe3O4@PMO (d–f) nanocomposites. | ||
Following the “in situ etching-growth” synthesis strategy, the PMO shells asymmetrically grow around the solid–liquid interface while the non-porous silica shells of the Fe3O4@nSiO2 cores are etched gradually. However, the self-assembly process of the organosilane oligomer/surfactant micelles is quite different from the former one. At a lower alkalinity, the homogenous growth of the radical PMO shells is interrupted, resulting in the asymmetrical growth of the cubic mesoporous PMO components and yielding a controllable void space (Fig. 4a, process I). In fact, the CTAB-organosilica micelles trend to grow into PMO crystals with a cubic mesostructure at a low alkalinity and PMO spheres with radical mesopore channels at a high alkalinity, even without the addition of the core materials (Fig. S15†). The magnetic PMO crystals with a hollow interior might serve as an ideal support for the storage, delivery and controlled release of guest molecules,44 and further take new opportunity in the assembly process.55
Compared to the methods reported previously for the preparation of nanocomposites with yolk–shell structure, our “in situ etching-growth strategy” has several advantages. Firstly, it combines the etching process and shell growth in one procedure, i.e., the condensed inner silica shells of the cores are etched and the outer PMO shells are constructed at the same time, so that the synthesis procedure is more facile for the fabrication of yolk–shell nanocomposites with mesoporous organosilica shells. Secondly, the size of void spaces can be well controlled by tuning the thickness of the condensed silica layers, or varying the time interval between the injection of silane and NaOH. What's more, the method is also a general route for the synthesis of yolk–shell nanomaterials with different sized cores, chemical compositions and shapes (Fe3O4 and dense silica nanospheres, silver nanocubes), as well as the organic moieties (ethane- or benzene-bridged) of the mesoporous shell frameworks. Moreover, the mesostructure of the PMO shells can be well controlled with radical or cubic mesopore channels via tuning the alkalinity. By slightly varying the synthesis parameters, mesoporous composites with various yolk–shell, core–shell structures and radical mesochannels, as well as asymmetric mesoporous composites with hollow structure and cubic PMO component (Pmn symmetry) can be obtained in the same system.
The yolk–shell structured mesoporous composites can be used in various areas, such as confining nanoparticles (Au, Ag, Pd, Pt, etc.) in the hollow interior of the composites to catalyze other reactions such as Suzuki–Miyaura cross-coupling and alcohol oxidation,50,56 and serving as supports (the PMO component) for different kinds of catalysts such as MacMillan catalysts49 for the Diels–Alder cycloaddition or adsorbents57,58 for arsenic pollution and microwave (the magnetic Fe3O4 cores). Herein, gold nanoparticles (Au NPs) can be loaded into the yolk–shell YS-Fe3O4@PMO composites by salt impregnation.50 The YS-Fe3O4@Au@PMO particles are still discrete and uniform (Fig. S16†), and Au NPs with the size of ∼25 nm are successfully loaded into the void space as shown in the TEM image (Fig. 5a). The weight percent of gold in the final composites is measured by XPS to be about 0.5% (Fig. S17a†), much lower than that from EDS (∼3.2%), because of the depth limitation of the X-ray radiation. Two bands are observed from the high-resolution Au 4f7 XPS spectra (Fig. S17b†) at 84.7 and 87.9 eV, which can be assigned to be the signal of Au 4f7/2 and Au 4f5/2 respectively, indicating the formation of metallic gold. Au NPs can catalyze many reactions such as the reduction of aromatic nitro-compounds or the epoxidation of styrene.11,59 Here, the magnetic yolk–shell composite loaded with Au NPs are employed to catalyze the reduction of p-nitrophenol (4-NP) by NaBH4 (Fig. S18†). The reaction did not occur without catalysts loaded with Au NPs. UV-vis absorption change of the reaction mixture after the addition of the YS-Fe3O4@Au@PMO particles versus time is plotted in Fig. 5b. The absorption peak of 4-NP at around 400 nm decreases as the time increases, and at the same time the band at ∼300 nm gradually increases. The reaction can be completed at 8 min, as the UV-vis plot becomes very flat at around 400 nm, revealing that 4-NP is totally digested. This reaction can be supposed as pseudo-first-order kinetic reaction since NaBH4 is highly excess and BH4− concentration keep nearly constant throughout the reaction. The linear relation of ln(Ct/C0) versus time (Fig. 5c) is observed and the first-order kinetics can be further confirmed. The slope of the fitted straight line is calculated as 0.01197 s−1, which can be also considered as the rate constant (k) of the reaction. This is much larger than many catalysts with Au NPs loaded in the nanochannels of porous materials, and comparable to some pure metallic catalysts. Such an excellent catalytic performance can be mainly attributed to the hollow space, high surface area of the yolk–shell mesoporous composites and uniform PMO shells with unique perpendicular mesopore channels for high-rate mass diffusion (Fig. S18†). By the utilization of the magnetic essence (Fig. S3 and S4†), the catalysts can be easily recovered and reused. The k value of the second cycle is 1.002 × 10−2 s−1, significantly decreases, but that for the next several cycles maintains nearly unchanged (>95% of the first cycle) even after 10 cycles (Fig. 5d). Besides, we believe the magnetic composites can be also used in other areas such as arsenic pollution removal and microwave adsorption.
 | ||
| Fig. 5 Characterization and catalytic performance of the yolk–shell YS-Fe3O4@Au@PMO composites with Au-NPs loaded in the void space. HRTEM image (a) of the YS-Fe3O4@Au@PMO composites, time-dependent UV-vis absorption spectral changes of the reaction mixture (b), plot of ln(Ct/C0) versus time (c) and cyclic catalytic performance versus the YS-Fe3O4@Au@PMO catalysts (d). | ||
4. Conclusions
In summary, we have demonstrated a general “in situ etching and growth” strategy for the fabrication of the yolk–shell structured mesoporous nanocomposites with movable cores of different sizes, chemical compositions, and shapes, and PMO based outer shells with controlled structure as well as various bridged organic groups. The new strategy combines the etching process with the PMO shell growth. The size of the void space (0–80 nm) can be easily tuned by varying the thickness of the sacrificial silica layer of the cores, or tuning the time interval between the addition of organosilanes and NaOH. Asymmetric Fe3O4@PMO composites with hollow space and cubic mesoporous PMO shells (Pmn symmetry) can be obtained at a relatively low alkalinity. All the magnetic composites have uniform ordered mesostructures with well-arranged mesopores (radical or cubic), high surface areas (586–946 m2 g−1) and large pore volumes (0.52–0.86 cm3 g−1), and moreover they are magnetic separable, therefore meet the demand in desired applications. Au-NPs can be loaded into the void space of the yolk–shell YS-Fe3O4@PMO composites via a simple salt impregnation method and used to catalyze the reduction of 4-NP by NaBH4, showing ultrahigh efficiency (k = 0.01197 s−1) and recyclability (retain >95% efficiency after 10 cycles). We believe the core–shell, yolk–shell, single-hole and Janus magnetic PMO composites can find more applications in bio-related fields, self-assembly of colloidal particles and so on.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the State Key Basic Research Program of the PRC (2012CB224805, 2013CB934104), the NSF of China (21210004 and U1463206), Shanghai Sci. & Tech. Committee (14JC1400700). We extended our appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the 530 research group Project No. RGP-227.References
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- J. Liu, S. Y. Bai, H. Zhong, C. Li and Q. H. Yang, J. Phys. Chem. C, 2010, 114, 953–961 CAS.
- J. Liu, Q. H. Yang, L. Zhang, H. Q. Yang, J. S. Gao and C. Li, Chem. Mater., 2008, 20, 4268–4275 CAS.
- S. H. Liu and M. Y. Han, Chem.–Asian J., 2009, 5, 36–45 Search PubMed.
- H. C. Zeng, J. Mater. Chem., 2006, 16, 649–662 RSC.
- Y. H. Deng, Y. Cai, Z. K. Sun and D. Y. Zhao, Chem. Phys. Lett., 2011, 510, 1–13 CrossRef CAS.
- J. Liu, S. Z. Qiao, Q. H. Hu and G. Q. Lu, Small, 2011, 7, 425–443 CrossRef CAS PubMed.
- Y. H. Deng, D. W. Qi, C. H. Deng, X. M. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- Z. K. Sun, Q. Yue, Y. Liu, J. Wei, B. Li, S. Kaliaguine, Y. H. Deng, Z. X. Wu and D. Y. Zhao, J. Mater. Chem. A, 2014, 2, 18322–18328 CAS.
- C. Wang, J. C. Chen, X. R. Zhou, W. Li, Y. Liu, Q. Yue, Z. T. Xue, Y. H. Li, A. A. Elzatahry, Y. H. Deng and D. Y. Zhao, Nano Res., 2015, 8, 238–245 CrossRef CAS.
- J. W. Liu, J. J. Xu, R. C. Che, H. J. Chen, M. M. Liu and Z. W. Liu, Chem.–Eur. J., 2013, 19, 6746–6752 CrossRef CAS PubMed.
- M. Lattuada and T. A. Hatton, Nano Today, 2011, 6, 286–308 CrossRef CAS.
- J. Yan, M. Bloom, S. C. Bae, E. Luijten and S. Granick, Nature, 2012, 491, 578–581 CrossRef CAS PubMed.
- J. H. Gao, G. Liang, B. Zhang, Y. Kuang, X. Zhang and B. Xu, J. Am. Chem. Soc., 2007, 129, 1428–1433 CrossRef CAS PubMed.
- W. R. Zhao, H. R. Chen, Y. S. Li, L. Li, M. D. Lang and J. L. Shi, Adv. Funct. Mater., 2008, 18, 2780–2788 CrossRef CAS.
- H. X. Li, Z. F. Bian, J. Zhu, Y. N. Huo, H. X. Li and Y. F. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed.
- K. Kamata, Y. Lu and Y. N. Xia, J. Am. Chem. Soc., 2003, 125, 2384–2385 CrossRef CAS PubMed.
- K. Zhang, X. H. Zhang, H. T. Chen, X. Chen, L. L. Zheng, J. H. Zhang and B. Yang, Langmuir, 2004, 20, 11312–11314 CrossRef CAS PubMed.
- P. Valle-Vigon, M. Sevilla and A. B. Fuertes, Mater. Lett., 2010, 64, 1587–1590 CrossRef CAS.
- X. W. Lou and L. A. Archer, Adv. Mater., 2008, 20, 1853–1858 CrossRef CAS.
- J. Kim, D. R. Arifin, N. Muja, T. Kim, A. A. Gilad, H. Kim, A. Arepally, T. Hyeon and J. W. M. Bulte, Angew. Chem., Int. Ed., 2011, 50, 2317–2321 CrossRef CAS PubMed.
- D. H. Park, J. E. Kim, J. M. Oh, Y. G. Shul and J. H. Choy, J. Am. Chem. Soc., 2010, 132, 16735–16736 CrossRef CAS PubMed.
- D. Chen, L. L. Li, F. Q. Tang and S. Qi, Adv. Mater., 2009, 21, 3804–3807 CrossRef CAS.
- Y. F. Zhu, E. Kockrick, T. Ikoma, N. Hanagata and S. Kaskel, Chem. Mater., 2009, 21, 2547–2553 CrossRef CAS.
- Y. Fang, G. F. Zheng, J. P. Yang, H. S. Tang, Y. F. Zhang, B. Kong, Y. Y. Lv, C. J. Xu, A. M. Asiri, J. Zi, F. Zhang and D. Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 5366–5370 CrossRef CAS PubMed.
- Y. Chen, H. R. Chen, L. M. Guo, Q. J. He, F. Chen, J. Zhou, J. W. Feng and J. L. Shi, ACS Nano, 2010, 4, 529–539 CrossRef CAS PubMed.
- T. R. Zhang, J. P. Ge, Y. X. Hu, Q. Zhang, S. Aloni and Y. D. Yin, Angew. Chem., Int. Ed., 2008, 47, 5806–5811 CrossRef CAS PubMed.
- S. J. Ding, J. S. Chen, G. G. Qi, X. N. Duan, Z. Y. Wang, E. P. Giannelis, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 21–23 CrossRef CAS PubMed.
- B. Liu and H. C. Zeng, Small, 2005, 1, 566–571 CrossRef CAS PubMed.
- Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorijai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- C. Urata, H. Yamada, R. Wakabayashi, Y. Aoyama, S. Hirosawa, S. Arai, S. Takeoka, Y. Yamauchi and K. Kuroda, J. Am. Chem. Soc., 2011, 133, 8102–8105 CrossRef CAS PubMed.
- J. Croissant, X. Cattoën, M. W. C. Man, A. Gallud, L. Raehm, P. Trens, M. Maynadier and J. Durand, Adv. Mater., 2014, 26, 6174–6180 CrossRef CAS PubMed.
- A. Zamboulis, N. Moitra, J. J. E. Moreau, X. Cattoën and M. W. C. Man, J. Mater. Chem., 2010, 20, 9322–9338 RSC.
- C. X. Lin, S. Z. Qiao, C. Z. Yu, S. Ismadji and G. Q. Lu, Microporous Mesoporous Mater., 2009, 117, 213–219 CrossRef CAS.
- V. Rebbin, R. Schmidt and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 5210–5214 CrossRef CAS PubMed.
- W. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2634–2642 CrossRef CAS PubMed.
- X. J. Wu and D. S. Xu, J. Am. Chem. Soc., 2009, 131, 2774–2775 CrossRef CAS PubMed.
- J. Liu, S. Z. Qiao, S. B. Hartono and G. Q. Lu, Angew. Chem., Int. Ed., 2010, 49, 4981–4985 CrossRef CAS PubMed.
- L. Zhang, S. Z. Qiao, Y. G. Jin, Z. G. Chen, H. C. Gu and G. Q. Lu, Adv. Mater., 2008, 20, 805–809 CrossRef CAS.
- X. M. Li, L. Zhou, Y. Wei, A. M. El-Toni, F. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2014, 136, 15086–15092 CrossRef CAS PubMed.
- S. Haffer, M. Tiemann and M. Froba, Chem.–Eur. J., 2010, 16, 10447–10452 CrossRef CAS PubMed.
- Y. Yang, J. Liu, X. B. Li, X. Liu and Q. H. Yang, Chem. Mater., 2011, 23, 3676–3684 CrossRef CAS.
- W. Na, Q. Wei and Z. R. Nie, J. Mater. Chem., 2012, 22, 9970–9974 RSC.
- J. Li, Y. Wei, W. Li, Y. H. Deng and D. Y. Zhao, Nanoscale, 2012, 4, 1647–1651 RSC.
- J. Y. Shi, C. A. Wang, Z. J. Li, Q. Wang, Y. Zhang and W. Wang, Chem.–Eur. J., 2011, 17, 6206–6213 CrossRef CAS PubMed.
- J. Liu, H. Q. Yang, F. Kleitz, Z. G. Chen, T. Y. Yang, E. Strounina, G. Q. Lu and S. Z. Qiao, Adv. Funct. Mater., 2012, 22, 591–599 CrossRef CAS.
- J. Liu, Z. K. Sun, Y. H. Deng, Y. Zou, C. Y. Li, X. H. Guo, L. Q. Xiong, Y. Gao, F. Y. Li and D. Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 5875–5879 CrossRef CAS PubMed.
- Q. Zhang, C. Cobley, L. Au, M. Mckiernan, A. Schwartz, L. P. Wen, J. Y. Chen and Y. N. Xia, ACS Appl. Mater. Interfaces, 2009, 1, 2044–2048 CAS.
- J. P. Yang, D. K. Shen, L. Zhou, W. Li, X. M. Li, C. Yao, R. Wang, A. M. El-Toni, F. Zhang and D. Y. Zhao, Chem. Mater., 2013, 25, 3030–3037 CrossRef CAS.
- H. Djojoputro, X. F. Zhou, S. Z. Qiao, L. Z. Wang, C. Z. Yu and G. Q. Lu, J. Am. Chem. Soc., 2006, 128, 6320–6321 CrossRef CAS PubMed.
- J. Yan, K. Chaudhary, S. C. Bae, J. A. Lewis and S. Granick, Nat. Commun., 2013, 4, 1516–1524 CrossRef PubMed.
- D. K. Shen, L. Chen, J. P. Yang, R. Y. Zhang, Y. Wei, X. M. Li, W. Li, Z. K. Sun, H. W. Zhu, A. M. Abdullah, A. Al-Enizi, A. A. Elzatahry, F. Zhang and D. Y. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 17450–17459 CAS.
- K. Folens, K. Leus, N. R. Nicomel, M. Meledina, S. Turner, G. V. Tendeloo, G. D. Laing and P. V. D. Voort, Eur. J. Inorg. Chem., 2016 DOI:10.1002/ejic.201600160.
- J. W. Liu, J. J. Xu, R. C. Che, H. J. Chen, M. M. Liu and Z. W. Liu, Chem.–Eur. J., 2013, 19, 6746–6752 CrossRef CAS PubMed.
- L. F. Tan, D. Chen, H. Y. Liu and F. Q. Tang, Adv. Mater., 2010, 22, 4885–4889 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The SEM and TEM images, small-angle and wide-angle XRD patterns, magnetic hysteresis loops, 29Si MAS-NMR, and nitrogen adsorption–desorption isotherms of the core materials and yolk–shell PMO composites, the XPS spectra of the YS-Fe3O4@Au@PMO composites were listed. See DOI: 10.1039/c6ra08541e |
| This journal is © The Royal Society of Chemistry 2016 |