DOI:
10.1039/C6RA08513J
(Paper)
RSC Adv., 2016,
6, 54898-54903
The effect of gold nanoparticles on the diagnostic polymerase chain reaction technique for equine herpes virus 1 (EHV-1)
Received
2nd April 2016
, Accepted 23rd May 2016
First published on 24th May 2016
Abstract
Nano-biotechnology has been a noticeable research area because of its successful applications in molecular diagnostics and therapy of various genetic and microbial diseases. Although the polymerase chain reaction (PCR) technique is one of the most highlighted and promising applications in the molecular diagnosis field, it suffers from some drawbacks that affect its efficiency. For instance, as a diagnostic technique for equine herpes virus-1 (EHV-1), conventional PCR could lead to false negative results due to the low viral titer in some samples, which leads to the necessity to improve its sensitivity. In this study, we carried out experiments to determine the effects of 15 nm unmodified citrate-coated gold nanoparticles (GNPs) on the key PCR reactants in order to see if these would enhance the overall outcomes of the reaction. Our results showed that, after optimization of the GNPs, oligonucleotide primers and Taq polymerase concentrations, a specific high yield amplification with a detection limit of 102 DNA copies could be reached compared to the 105 to 104 detection limit of conventional PCR. Thus, the developed and optimized GNPs-assisted PCR technique could be used for a more efficient, highly sensitive molecular detection of EHV-1.
1. Introduction
The polymerase chain reaction (PCR) is a breakthrough technique that was first explored and improved by Kary Mullis in 1983.1,2 It employs an in vitro DNA amplification system that mimics the in vivo DNA replication by the simultaneous primer extension of the complementary strands of DNA based on a set of repeated temperature cycles.3 Nowadays, it has become one of the most important and reliable techniques in molecular diagnosis,4 biotechnology, cloning,5 fingerprinting,6 microarray7 and genetic analysis8 fields. Although it is a very sensitive technique, it suffers from some specificity and efficiency drawbacks9,10 as it constitutes an error-prone in vitro reaction that lacks an in vivo DNA replication control mechanisms, such as the single stranded DNA binding protein (SSBP), which results in a highly specific satisfactory replication.11 To overcome these problems, some additives are added to the reaction mixture, such as dimethylsulfoxide12 (DMSO), amidoamines,13 bovine serum albumin14 (BSA), dithiotheritol14 (DTT) and glycerol.14 Furthermore, novel modifications in PCR techniques have been developed to accomplish higher reaction efficiency, e.g. hot start PCR15 and touchdown PCR.16
The evolution of the nanotechnology field since 1980 and interdisciplinary applied research have led to great applications in molecular biology and biotechnology. Recently, the addition of different types of nanoparticles to PCR has attracted significant attention due to their desirable chemical and physical properties.17,18 Nanoparticle-assisted PCR, or the nano-PCR technique, has been investigated for enhancing the efficiency of the PCR technique.
Gold nanoparticles (GNPs) were first introduced as a new additive in PCR to avoid non-specificity even at lowered annealing temperatures19 and to enhance the overall reaction. There are two hypotheses that exist that might explain how the enhancement occurs in PCR when GNPs are added: the first suggests that the enhancement occurs due to the surface interactions of the GNPs with the PCR reactants, while the other suggests that the enhancement occurs due to the heat transfer by the high conductivity and high heat ability of GNPs.19,20 Until now the actual mechanism of the enhancement still a mystery under investigation. However, in related studies, it was reported that GNPs-assisted PCR could be used for the more sensitive diagnosis of various disease causative agents, such as some bacteria21 and viruses.22–24
Equine herpes virus 1 (EHV-1) is a DNA virus that infects the equine species, with a worldwide distribution and where the infection can be subclinical or can cause serious economic loss in the equine industry as can cause sporadic abortion, neonatal death, equine herpes myeloencephalopathy (EHM) and upper respiratory diseases.25
In the present study, we highlight the effects of 15 nm unmodified citrate-coated GNPs on the key components of the diagnostic PCR technique for EHV-1. The sensitivity of the developed GNPs-assisted PCR was determined in comparison to the conventional PCR technique in order to achieve a more efficient diagnostic tool for EHV-1.
2. Experimental section
2.1 Reagents
Hydrogen tetrachloroaurate(III)trihydrate and trisodium citrate dihydrate were purchased from Alfa Aesar (UK). EHV-1 vaccine was supplied from Duvaxyn® (Australia). EHV-1 oligonucleotide primers were purchased from Metabion (Germany). GoTaq® G2 Flexi DNA polymerase was purchased from Promega (USA). Agarose, Tris base, boric acid and EDTA were purchased from Sigma Aldrich (USA). GeneJET Genomic DNA Purification Kit was purchased from Thermo Scientific™ (Germany). Ethidium bromide was supplied from Serva Electrophoresis (USA) and DNA marker from GeneDirex® (USA).
2.2 GNPs synthesis
Gold nanoparticles were synthesized using the improved Turkevich–Frens synthesis method.26 Briefly, after boiling an aqueous solution of 0.02% HAuCl4 (50 mL) with rapid stirring, 5% sodium citrate (1.176 mL) was added under lower stirring velocity. The colour of the aqueous solution changes from pale yellow to grey, black, deep violet and finally to ruby red. After reaching the ruby red colour, the solution was boiled for an additional 2 minutes to ensure that there is no further colour change, then it was allowed to cool down to room temperature. The synthesized GNPs were characterized by a JEOL-JEM-2100 Transmission Electron Microscope (TEM) and BMG SPECTROstar Nano UV-visible spectrometer.
2.3 Preparation of DNA template
EHV-1 vaccine DNA was used as a template for the PCR techniques. The extraction of viral DNA was done using the GeneJET Genomic DNA Purification Kit according to the supplier manual. The DNA was eluted with 50 μL of elution buffer and its concentration and purity were measured with a BMG SPECTROstar Nano UV-visible spectrometer; then it was stored at −20 °C for further use.
2.4 Establishment of conventional PCR for EHV-1 diagnosis
To amplify the 188 pb species-specific fragment for EHV-1, a pair of oligonucleotide primers specific for glycoprotein B gene (Table 1) was used. PCR was carried out in a 15 μL reaction mixture composed of 5× Taq DNA polymerase buffer (3.3 μL), 10 mmol dNTPs (0.6 μL), Taq DNA polymerase (1.25 units), 10 pmol primers mix (1 μL) and DNA template (1 μL), then the reaction was completed to 15 μL with PCR grade water. The PCR program was run as follows: initial denaturation step at 94 °C for 5 min, then 35 cycles of: denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s and extension at 72 °C for 30 s; then, a final extension at 72 °C for 5 min. 6 μL of PCR product was loaded and migrated onto 1.5% agarose gel containing 0.5 pg of ethidium bromide dye. The gel electrophoresis was run under constant voltage (85 V) for 45 min. After electrophoresis, the gel was visualized, photographed and analysed by Bio-Rad Gel Doc XR documentation system. The size of the resulted bands was evaluated compared to 50 bp DNA markers. For the optimization of this conventional PCR, different annealing temperatures ranging from 55 to 60 °C and different primer concentrations ranging from 0.33 to 1.67 μM were used.
Table 1 Oligonucleotide primers used for EHV-1 specific fragment amplification by PCR
| Name |
Sequence (5′-3′) |
Location |
Product size |
Ref. |
| FC3 |
ATACGATCACATCCAATCCC |
gB 2699–2718 |
188 pb |
27 |
| R1 |
GCGTTATAGCTATCACGTCC |
gB 2886–2867 |
2.5 GNPs-assisted PCR optimization
2.5.1 GNPs concentration optimization. Different concentrations of GNPs (0.2, 0.4, 0.6, 0.8, 1.0, 1.5 and 2 nM) were introduced to the original PCR reaction under the same conditions and PCR program to highlight the general effect of GNPs on PCR yield and sensitivity. The PCR products were analyzed and photographed as previously described.
2.5.2 Primer, Taq polymerase and BSA concentrations optimization. Different primers, Taq polymerase and BSA concentrations varied from 0.167–1.67 μM, 1.25–7 units, 0.2–1 μg μL−1, respectively, were added to the reaction after the optimization of the GNPs concentrations. The reaction was carried out using the same PCR program to estimate the effect of GNPs on each reactant. The PCR products were analyzed and photographed as previously described.
2.5.3 Sensitivity. To compare the sensitivity of GNPs-assisted PCR and conventional PCR, 10-fold serial dilutions of the template EHV-1 DNA were used.
2.6 PCR product quantification
Bio-Rad Gel Doc XR documentation system software was used to quantify each band intensity resulting from the migration of the amplified PCR product on 1.5% agarose gel. The measured band intensities were expressed in adjusted volumes (Int.).
3. Results and discussion
3.1 Characterization of the synthesized gold nanoparticles
GNPs sized 15 ± 3 nm were produced by the improved Turkevich–Frens method. The synthesized GNPs were indicated by the appearance of a ruby red colour. The TEM image showed that the synthesized GNPs had a relatively regular size with a spherical morphology (Fig. 1A). The UV-vis spectroscopy showed a λmax specific peak at a wavelength of 521 nm (Fig. 1B).
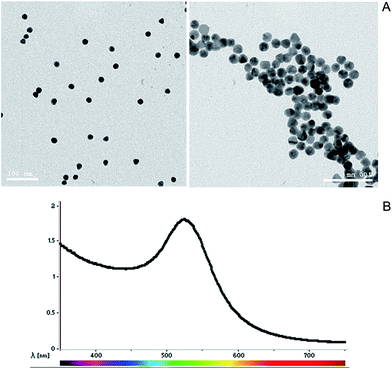 |
| | Fig. 1 (A) TEM image of the synthesized GNPs with a relatively regular size and spherical morphology. (B) UV-visible spectroscopy specific peak at a wavelength of 521 nm. | |
3.2 Optimization of the conventional PCR technique
For better PCR outcome, the PCR conditions were optimized relative to the annealing temperature and primer concentration. The effect of these factors was determined by the band intensity of the migrated PCR product on 1.5% agarose gel. The highest PCR yield was detected at an annealing temperature of 60 °C (Fig. 2A) and a primer concentration of 0.67 μM (Fig. 2B).
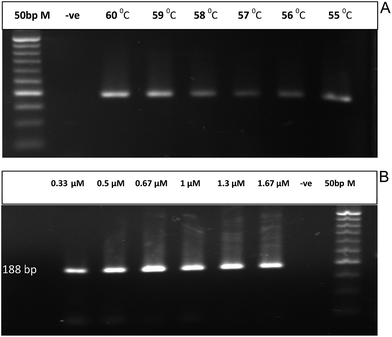 |
| | Fig. 2 (A) Conventional PCR amplification of 188 bp specific fragment for EHV-1 at different annealing temperatures ranging from 55 to 60 °C and compared to a negative control (−ve) and 50 bp marker. (B) Conventional PCR amplification of 188 bp specific fragment for EHV-1 at different primer concentrations ranging from 0.33 to 1.67 μM and compared to a negative control (−ve) and 50 bp marker. | |
3.3 Effect of different GNPs concentrations on conventional PCR
To determine the best GNPs concentration that give the highest PCR yield, PCR was carried out with different GNPs concentrations ranging from 0.2 to 2 nM. The PCR yield is directly proportional to the intensity of PCR product band on the agarose gel. It was found that the optimal GNPs concentration in the PCR reaction was 1 nM, which had a 3-fold higher band intensity compared to a reaction without the addition of any GNPs (Fig. 3). The intensity of the band increases as the concentration of the GNPs increases up to 1 nM, then it starts to decline with higher GNPs concentrations such as 1.5 and 2 nM. Although the interaction between GNPs and the PCR reactants is not fully clarified yet, this enhancement may be due to: (1) the GNPs act like SSBP that is present in the DNA replication system, which increases the specificity; (2) the GNPs regulate PCR through their interaction with the polymerase enzyme; (3) the GNPs enhance PCR specificity through the adjustment of Tm in the annealing step; (4) the GNPs increase the product yield through allowing the efficient dissociation of the PCR products in the denaturation step. Hence, there is more DNA template for more PCR products.28 It was found that an excess concentration of GNPs in the reaction could decrease its efficiency or even cause a total inhibition of the reaction.19 In related studies, it was revealed that the total surface area of the GNPs is the main cause in the inhibition rather that their size.29,30 The inhibition effect can occur with high concentration of GNPs where DNA polymerase can be strongly but not completely adsorbed on the negatively charged surface of GNPs, with their polar groups modulating the amount of active polymerases in the reaction. By this concept, GNPs will lower the PCR efficiency. This effect could be overcame by using higher concentrations of Taq polymerase or competitive proteins, such as BSA and thrombin, where both compete with Taq polymerase to bind on the GNPs surface through a Vroman-like effect.31,32 These effects of GNPs on the PCR technique basically depend on the surface interaction with different reaction components, such as Taq polymerase, primers and products, where they are kinetically adsorbed and dissociated from the GNPs surface.33 For more clarification on the effect of GNPs on PCR, more studies were performed on different PCR reactants.
 |
| | Fig. 3 The effects of different GNPs concentrations on PCR amplification of 188 bp specific fragment for EHV-1 compared to a reaction without (W/O) the addition of GNPs and 50 bp marker (A). Relative quantification of the PCR yield through measuring the band intensity (B). | |
3.4 Effect of GNPs on primer concentration
After the optimization of GNPs concentration in the PCR mix, we tested different concentrations of PCR primers, ranging from 0.167–1.67 μM, to highlight the effect of GNPs on the PCR buffer and primers. Fig. 4 shows that the optimal primer concentration to increase the PCR yield and specificity is 1 μM. Oligonucleotide primers, as short negatively charged DNA strands, interact with the GNPs surface due to adsorption–desorption kinetics as they replace the negatively charged citrate ions on the surface of GNPs. These single stranded DNA (ssDNA) sequences attach much more strongly than double stranded DNA (dsDNA) sequences to GNPs because of the easier bases exposure to the GNPs surface. The PCR primers attach to GNPs until they associate with their complementary sequences on the DNA template.34 This adsorption causes a decrease of the melting temperatures (Tm) for both complementary and mismatched primers and increases the Tm difference between them. This may explain the improved specificity and yield of GNPs-assisted PCR. PCR buffer usually contains NaCl and MgCl2, which are considered to cause GNPs aggregation, unless a certain amount of ssDNA is added into the reaction.35 GNPs effects differ from one assay to another according to the different binding affinities of the nucleotides towards the GNPs surface, where adenine shows a higher binding affinity for GNPs surfaces than thymine, while guanine and cytosine show a similar but less affinity to the GNPs surface.
 |
| | Fig. 4 The effect of different primer concentrations on GNPs-assisted PCR amplification of 188 bp specific fragment for EHV-1 compared to 50 bp marker (A). Relative quantification of the PCR yield through measuring the band intensity (B). | |
3.5 Effect of GNPs on Taq polymerase
To evaluate the effect of GNPs on Taq polymerase, PCR was carried out using 1 nM of GNPs and different Taq polymerase concentrations ranging from 1.25 to 7 units. Fig. 5 shows that the optimal Taq polymerase concentration is 4.25 units. PCR yield increases as the units of Taq polymerase increases up to 4.25 units, then it declines with higher concentrations. This effect is due to the fact that an excessive concentration of Taq polymerase leads to an increase in the artefacts associated with its intrinsic exonuclease activity, resulting in producing unwanted DNA fragments or complete reaction inhibition.36 The gradual increase in PCR yield up to 4.25 units was due to the compensation of the added Taq polymerase units for the modulated amount of Taq polymerase by GNPs. PCR efficiency enhancement by GNPs mostly depends on the type of polymerase used in the reaction.37
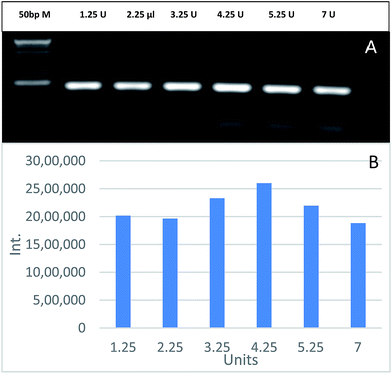 |
| | Fig. 5 The effects of different Taq polymerase concentrations on GNPs-assisted PCR amplification of 188 bp specific fragment for EHV-1 compared to 50 bp marker (A). Relative quantification of the PCR yield through measuring the band intensity (B). | |
3.6 Effect of GNPs on PCR containing BSA
PCR was carried out using 1 nM of GNPs and different BSA concentrations. Fig. 6 shows that there is no significant change in the band intensity of the PCR products between the GNPs-assisted PCR reaction without BSA and with different concentrations of BSA ranging from 0.2 to 1 μg μL−1. It is well known that BSA can improve the efficiency of conventional PCR.14 It was hypothesized that BSA could be used to modulate the adsorption of Taq polymerase and act as a competitor for the GNPs surface to increase the efficiency of GNPs-assisted PCR. In this study, we found that the addition of BSA to GNPs-assisted PCR had very little effect on the PCR yield. This effect could be explained as the BSA is a protein with a low isoelectric point (pI = 4.7) compared to Taq polymerase (pI = 6), while the typical PCR mix has a pH ∼ 8.3, which means that BSA retains more negative charges than Taq polymerase, leading to stronger repulsion with the negatively charged GNPs and DNA. Thus, less efficient adsorption on the surface of GNPs.35
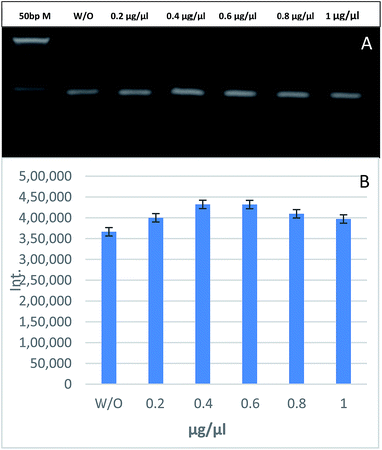 |
| | Fig. 6 The effects of different BSA concentrations on GNPs-assisted PCR amplification of 188 bp specific fragment for EHV-1 compared to 50 bp marker (A). Relative quantification of the PCR yield through measuring the band intensity (B). | |
3.7 Sensitivity of GNPs-assisted PCR vs. conventional PCR
To examine the GNPs-assisted PCR sensitivity, 10-fold serial dilutions of the template EHV-1 DNA were added to the PCR mixture containing 1 nM of GNPs. Fig. 7 shows a comparison between the sensitivity of GNPs-assisted PCR (A) and the sensitivity of conventional PCR (B), where a serial dilution of DNA template from 107 to 101 was used. The detection limit of the GNPs-assisted PCR is 102, which means that it can detect 100 copies of the target DNA, while the detection limit of the conventional PCR is 104, as determined through the experimental repetition. If the UV-trans illuminator is used to visualize the gel, the 104 dilution band cannot be clearly seen as a positive band and it needs the high resolution CCD camera of the gel documentation system to be recognized.
 |
| | Fig. 7 Sensitivity of GNPs-assisted PCR (A) versus conventional PCR (B). Serial dilution of 10-fold of EHV-1 DNA was used. All experiments underwent a 3 times repetition and similar results were achieved. Relative quantification of the PCR yield through measuring the band intensity, for the serial dilution of the GNPs-assisted PCR (C) and the serial dilution of the conventional PCR (D). | |
4. Conclusion
Every reactant added into the PCR mixture, including primers, DNA polymerase enzyme and BSA concentrations as well as DNA template and primer sequences, and also the size and surface modification of GNPs all affect the PCR efficiency. Consequently, it is important to evaluate these factors on a case-by-case basis as it will provide different results. In this study, the PCR technique was dramatically enhanced after the optimization of the GNPs, PCR primers and Taq polymerase concentrations to 1 nM, 1 μM and 4.25 U, respectively. It was found that BSA had very little effect on the GNPs-assisted PCR technique. Moreover, the higher sensitivity of the GNPs-assisted PCR technique, with a detection limit of 102 DNA copies, allowed the detection of lower viral titer and therefore detection of the virus at an early stage of the infection. It also eliminates the necessity to carry out the two round PCR (nested PCR). We found that the developed GNPs-assisted PCR technique could be used as a highly efficient, sensitive and specific diagnostic technique for EHV-1 rather than conventional PCR.
References
- K. Mullis, F. Faloona, S. Scharf, R. Saiki, G. Horn and H. Erlich, Cold Spring Harbor Symp. Quant. Biol., 1986, 51, 263–273 CrossRef CAS PubMed.
- J. M. S. Bartlett and D. Stirling, Methods Mol. Biol., 2003, 226, 3–6 CAS.
- M. Joshi and J. D. Deshpande, Int. J. Biomed. Res., 2010, 1, 81–97 Search PubMed.
- S. Yang and R. E. Rothman, Lancet Infect. Dis., 2004, 4, 337–348 CrossRef CAS PubMed.
- J. N. Larsen, P. Strøman and H. Ipsen, Mol. Immunol., 1992, 29, 703–711 CrossRef CAS PubMed.
- J. Welsh and M. McClelland, Nucleic Acids Res., 1990, 18, 7213–7218 CrossRef CAS PubMed.
- M. Huber, A. Mündlein, E. Dornstauder, C. Schneeberger, C. B. Tempfer, M. W. Mueller and W. M. Schmidt, Anal. Biochem., 2002, 303, 25–33 CrossRef CAS PubMed.
- J. Shendure and H. Ji, Nat. Biotechnol., 2008, 26, 1135–1145 CrossRef CAS PubMed.
- R. K. Agarwal and A. Perl, Nucleic Acids Res., 1993, 21, 5283–5284 CrossRef CAS PubMed.
- E. Ashrafi and N. Paul, BioTechniques, 2009, 47, 789–790 CrossRef CAS PubMed.
- R. R. Meyer and P. S. Laine, Microbiol. Rev., 1990, 54, 342–380 CAS.
- J. Kang, M. S. Lee and D. G. Gorenstein, J. Biochem. Biophys. Methods, 2005, 64, 147–151 CrossRef CAS PubMed.
- X. Cao, X. Shi, W. Yang, X. Zhang and J. Hu, Analyst, 2009, 87–92 RSC.
- N. S. Masazumi Nagai and A. Yoshida, Biochem. Mol. Biol. Int., 1998, 44, 157–163 Search PubMed.
- A. V. Lebedev, N. Paul, J. Yee, V. A. Timoshchuk, J. Shum, K. Miyagi, J. Kellum, R. I. Hogrefe and G. Zon, Nucleic Acids Res., 2008, 36, 1–18 CrossRef PubMed.
- D. J. Korbie and J. S. Mattick, Nat. Protoc., 2008, 3, 13–15 Search PubMed.
- C. Binns, Introduction to Nanoscience and Nanotechnology, 2010 Search PubMed.
- J. L. Feather and M. F. Aznar, Principles, Applications, Implications and Hands-on Activities, 2010 Search PubMed.
- H. Li, J. Huang, J. Lv, H. An, X. Zhang, Z. Zhang, C. Fan and J. Hu, Angew. Chem., Int. Ed., 2005, 44, 5100–5103 CrossRef CAS PubMed.
- S. Mandal, M. Hossain, T. Muruganandan, G. S. Kumar and K. Chaudhuri, RSC Adv., 2013, 3, 20793 RSC.
- A. Rehman, Y. Sarwar, Z. A. Raza, S. Z. Hussain, T. Mustafa, W. S. Khan, M. A. Ghauri, A. Haque and I. Hussain, Analyst, 2015, 140, 7366–7372 RSC.
- J. Wang, Y. Cheng, M. Zhang, H. Zhao, P. Lin, L. Yi, M. Tong and S. Cheng, BMC Vet. Res., 2015, 11, 1 CrossRef PubMed.
- W. Yuan, Y. Li, P. Li, Q. Song, L. Li and J. Sun, J. Virol. Methods, 2015, 220, 18–20 CrossRef CAS PubMed.
- X. Wang, A. Bai, J. Zhang, M. Kong, Y. Cui, X. Ma, X. Ai, Q. Tang and S. Cui, J. Virol. Methods, 2014, 202, 106–111 CrossRef CAS PubMed.
- M. Dunowska, N. Z. Vet. J., 2014, 62, 179–188 CrossRef CAS PubMed.
- K. Zabetakis, W. E. Ghann, S. Kumar and M. C. Daniel, Gold Bull., 2012, 45, 203–211 CrossRef CAS.
- L. Wang, S. L. Raidal, A. Pizzirani and G. E. Wilcox, Vet. Microbiol., 2007, 121, 18–28 CrossRef CAS PubMed.
- Y. Lin, J. Li, J. Yao, Y. Liang, J. Zhang, Q. Zhou and G. Jiang, Chin. Sci. Bull., 2013, 58, 4593–4601 CrossRef CAS.
- B. V. Vu, D. Litvinov and R. C. Willson, Anal. Chem., 2008, 80, 5462–5467 CrossRef CAS PubMed.
- D. Pan, Y. Wen, L. Mi, C. Fan and J. Hu, Curr. Org. Chem., 2011, 15, 486–497 CrossRef CAS.
- L.-P. Sun, S. Wang, Z.-W. Zhang, Y.-Y. Ma, Y.-Q. Lai, J. Weng and Q.-Q. Zhang, IET Nanobiotechnol., 2011, 5, 20 CrossRef CAS PubMed.
- A. L. Adams, Blood, 1980, 55, 156–159 Search PubMed.
- L. Mi, Y. Wen, D. Pan, Y. Wang, C. Fan and J. Hu, Small, 2009, 5, 2597–2600 CrossRef CAS PubMed.
- H. Li and L. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14036–14039 CrossRef CAS PubMed.
- X. Lou and Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 6276–6284 CAS.
- T. C. Lorenz, J. Vis. Exp., 2012, e3998 Search PubMed.
- M. Li, Nucleic Acids Res., 2005, 33, e184 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 






