
DFT study of ethanol dehydration catalysed by hematite†
Abstract
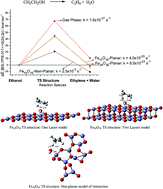
Hematite (α-Fe2O3) has been used as an ethanol gas sensor and as a catalyst for nanomaterial synthesis, motivating the investigation of ethanol dehydration over a hematite surface using computational chemistry methods. Using a molecular approach and a simple model for α-Fe2O3 to mimic the catalyst effect on the thermodynamics and kinetics of the reaction and quantum chemical Density Functional Theory (DFT) calculations, we showed that the energy barrier for the formation of ethylene plus water is around 70% lower than the corresponding gas phase value and that the products are thermodynamically stable with respect to the ethanol reactant in the presence of a non-planar model catalyst (Fe10O15). This stimulating result supports a mechanism proposed for ethylene formation at mild temperatures using hematite as a catalyst. We also showed that the effect of a catalyst on a chemical process can be very satisfactorily simulated as a local effect through first principles quantum chemical calculations, being of practical use, and so, large cluster calculations with many atomic layers or the use of large periodic super cells are not strictly required to reach reasonable estimates of temperature dependent rate constants and products stabilization. Our results strongly support a proposal that the main intermolecular interactions between the species present in a chemical process and the catalyst, leading to stabilization of transition state structure and consequent reduction of energy barrier can be fairly described by the DFT level of theory using small molecular models based on sound chemical ground.
 Please wait while we load your content...
Please wait while we load your content...

