
Influence of acid site density on the three-staged MTH induction reaction over HZSM-5 zeolite†
Abstract
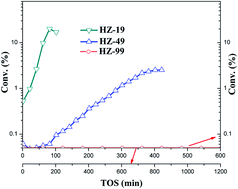
The influence of acid site density on the three-stage methanol to hydrocarbons (MTH) induction reaction was systematically investigated over three HZSM-5 catalysts (average size of 1–2 μm) with different Si/Al ratios. The initial active species can be formed more easily over catalysts with higher acid site density during the initial two stages and co-feeding toluene could shorten the initial two stages in the temperature-programmed MTH reaction. For each catalyst, the kinetics of the autocatalysis reaction during the induction period was investigated and the corresponding activation energy was calculated and compared. It was found that the formation rate of the retained species and its autocatalytic effect can both be enhanced over samples with high acid site density. Organic materials occluded in the catalyst during the MTH induction reaction were also analyzed ex situ by thermal analysis and GC-MS after extraction. The characterization results corresponded with the kinetic results very well.
 Please wait while we load your content...
Please wait while we load your content...

