
Crystal structure, thermal crystal form transformation, desolvation process and desolvation kinetics of two novel solvates of ciclesonide†
Abstract
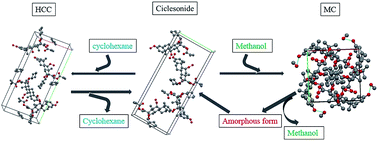
Two novel solvates of ciclesonide were successfully obtained and characterized by various analytical techniques (X-ray powder diffraction, X-ray single-crystal diffraction, differential scanning calorimetry, thermogravimetric analysis and hot-stage microscopy). Crystal structure analysis results indicate that the solvate of ciclesonide formed with cyclohexane (HCC) is a channel solvate and isostructural with ciclesonide, while the solvate formed with methanol (MC) isa discrete position solvate. Thermal analysis results show that HCC could desolvate without destroying the crystal lattice. However, MC would transform into an amorphous form after desolvation and the amorphous form could recrystallize into the crystalline form of ciclesonide. Moreover, the kinetics of the HCC and MC isothermal and non-isothermal desolvation processes is calculated and discussed.
 Please wait while we load your content...
Please wait while we load your content...

