
A new electrolyte with good compatibility to a lithium anode for non-aqueous Li–O2 batteries†
Abstract
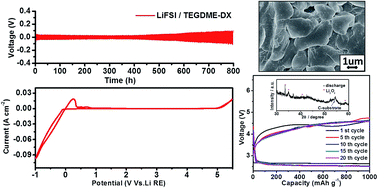
A novel LiFSI/TEGDME-DX electrolyte with good compatibility to a lithium anode is firstly proposed for the rechargeable non-aqueous Li–O2 battery, in which an improved performance with longer cycle life was achieved when compared with the conventional LiTFSI/TEGDME electrolyte.
 Please wait while we load your content...
Please wait while we load your content...

