
Isotopic homogenization and scrambling associated with oxygen isotopic exchange on hot platinum: studies on gas pairs (O2, CO2) and (CO, CO2)†
Abstract
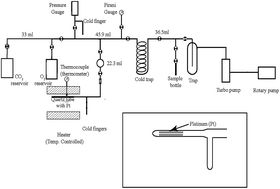
The catalytic exchange between O2 and CO2 on hot platinum leads to isotope scrambling in CO2 and homogenization of the oxygen isotopes in the two phases (O2 and CO2) even though the two gases could be widely different in isotope ratios from each other (e.g., about 45‰ in δ18O) before the exchange. After the exchange, the δ18O values of the gases become close to each other (within 2‰) depending on the time and temperature. The clumped-isotope analysis of the post-exchange CO2 samples shows that the Δ47 values (relative to the pre-exchange values) decrease with increase in temperature from near zero (at ∼25 °C) to about −0.8 ± 0.1‰ (at ∼700 °C). The low value of −0.8‰ equals the values typically obtained for pure CO2 heated to 1000 °C inside quartz tubes suggesting a scrambled state. If the catalytic exchange between CO2 and O2 is associated with isotopic scrambling (or random mixing) a microscopic mechanism of isotopic mixing can be inferred. It seems that CO2 and O2 dissociate in the adsorbed state on platinum (or quartz surfaces) and the product CO molecules and O atoms mix uniformly while doing a random walk on the hot surface and produce two reservoirs (CO and O) where the three oxygen isotopes are randomly distributed. This can produce the observed internal isotopic scrambling in CO2 when CO and O recombine to form CO2 molecules on the platinum surface and desorb. Experiments using CO–CO2 gas pairs over hot platinum also show homogenization showing exchange of CO as a molecular entity and supporting the suggested mechanism.
 Please wait while we load your content...
Please wait while we load your content...

