
Photothermal conversion upon near-infrared irradiation of fluorescent carbon nanoparticles formed from carbonized polydopamine†
Abstract
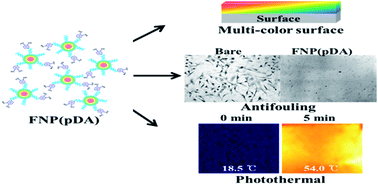
Fluorescence and photothermal conversion mediated by near-infrared radiation (NIR) is reported for carbonized polydopamine nanoparticles. Carbonized polydopamine demonstrated excitation-dependent fluorescence emission, together with NIR-responsive photothermal conversion properties. The concentration-dependent photothermal heating from carbonized fluorescent carbon nanoparticles-polydopamine (FNP-pDA) induces hyperthermal killing of both cancer cell lines and bacteria in vitro. Although most of the dopamine moieties of polydopamine become dehydrated upon carbonization, the remaining dopamine-hydroxyl groups can confer adhesive properties. These fluorescent coatings are compatible with many substrates, and the surface passivation of FNP-pDA with polyethylene glycol improves quantum yield and extends fluorescence lifetimes. The novel infrared-responsive photothermal and fluorescent carbon nanoparticles reported here show promise for a range of potential biomedical and research applications.
 Please wait while we load your content...
Please wait while we load your content...

