
Catalysis by multifunctional polyelectrolyte capsules†

Abstract
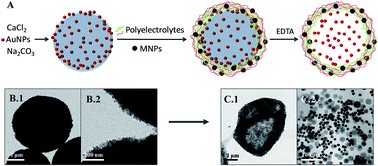
Gold nanoparticles and nanocomposites have high catalytic performance for several chemical reactions. Here we present gold and iron oxide nanoparticle modified polymer capsules as porous and multifunctional platforms for catalysis. Layer-by-layer polyelectrolyte microcapsules were formed on calcium carbonate template cores loaded with gold nanoparticles, allowing for high gold loading of the capsules. Magnetic nanoparticles were incorporated in the polymeric shells of the capsules, allowing for magnetic separation. The influence on the catalytic behaviour of gold was studied in terms of the nanoparticle size, the presence of a polymeric shell, and the presence of the magnetic nanoparticles in the shell, by using the model electron transfer reaction between hexacyanoferrate(III) and borohydride.
 Please wait while we load your content...
Please wait while we load your content...

