DOI:
10.1039/C6RA08111H
(Paper)
RSC Adv., 2016,
6, 78409-78423
Eco-friendly grinding synthesis of a double-layered nanomaterial and the correlation between its basicity, calcination and catalytic activity in the green synthesis of novel fused pyrimidines†
Received
29th March 2016
, Accepted 25th July 2016
First published on 25th July 2016
Introduction
Pyrimidine and its derivatives have been studied for several years because of their chemical and biological significance. They have been reported as antiviral, antitumour, anti-inflammatory, antihypertensive activities,1–3 calcium channel modulators,4 and antimicrobial agents.5–7 Numerous heterocyclic systems fused with pyrimidines are known for their important biological activities.8 Some chromenopyrimidine derivatives show antiplatelet and antithrombotic activities.9 They also exhibit various types of biological activity, including analgesic,10 antibiotic,11 cytotoxic12 and antitumour properties.13 The thiazole moiety is a prevalent scaffold in a number of naturally occurring and synthetic molecules with attractive biological activities, such as antiviral, anticancer, antibacterial, antifungal, anticonvulsant, antiparkinsonian and anti-inflammatory, that are well illustrated by the large number of drugs in the market containing this heterocyclic moiety.14–21 Coumarin derivatives, which are widely distributed in plants,22 have been extensively investigated as anticoagulation, antiviral,23 anti-inflammatory,24 antibacterial25 and anticancer agents.26
Layered double hydroxides (LDHs), which are referred to as anionic clays in comparison with cationic clays and also as hydrotalcite-like compounds (HT), are an important class of ionic lamellar solid. LDHs have the general formula [M(1−x)2+Mx3+(OH)2]y+[An−]y/n]·mH2O, where M2+ and M3+ are divalent (Mg2+, Zn2+, Ni2+, etc.) and trivalent cations (Al3+, Cr3+, etc.), and x is normally between 0.17 and 0.33.27 The main property of hydrotalcites is their anion exchange capacity, which makes them unique inorganic materials to intercalate organic or inorganic anions.28 Hydrotalcites are increasingly regarded as a good alternative to the traditional homogenous base catalysts, such as NaOH and KOH, for several base-catalyzed reactions that are important for the pharmaceutical and fragrance industries,29 as electrode modifiers and catalyst supports.30 A well-documented example is the isomerization of eugenol and safrole.31 Their structure consists of positively charged brucite (magnesium hydroxide)-like layers with an interlayer space containing charge compensating anions and water molecules.32 As far as green chemistry is concerned, hydrotalcites offer several advantages over the traditional corrosive, dissolved catalysts, such as easy separation from the reaction mixture, recycling possibilities and decreased corrosion of the reactor.33
The variety of applications of hydrotalcite-based materials is virtually unlimited. Hydrotalcites can be implicated in the preparation of catalysts dedicated to the production of H2,34 a wide range of organic compounds35 and the production of biodiesel by the trans-esterification of triglycerides with methanol.36 In addition to the above, numerous experimental investigations have been published on the use of hydrotalcites for catalytic applications.37 A study on the synthesis of hydrotalcites has received considerable attention, and to date, various methods have been reported, including the salt-oxide method,38 co-precipitation method,39 induced hydrolysis,40 reconstruction,41 anion-exchange42 sol–gel43 and hydrothermal treatment.44 Various authors43,45,46 reported a synthetic hydrotalcite preparation at high temperature (325–350 °C) and in a long duration synthesis (18 h). However, co-precipitation at a fixed pH is the most commonly used method. The purpose of the present work was to develop a new green method that is simple, more economical, free of impurities and industrially feasible. Thus, the present method fulfils the requirements of rapid synthesis and good quality.
Therefore, as a part of our ongoing research aimed at the development of new catalysts and their application in the synthesis of heterocycles,47 we report herein a rapid and cost-effective method (grinding method) for the synthesis of hydrotalcite, and furthermore, we demonstrate it in the synthesis of novel fused pyrimidines via a three-component reaction under solvent-free conditions.
Results and discussion
Characterization of the hydrotalcite
The IR spectra (Fig. 1) show absorption bands at higher wavenumbers of 3695 and 3471 cm−1, which are assigned to the O–H stretching vibrations of water bonded to M3OH units. The shorter O–H bonds existing in the hydrotalcite causes an increase in electrostatic attraction within the hydrotalcite layers.48 The weak bands at 2930 cm−1 and 2880 cm−1 suggest strongly hydrogen bonded water molecules to interlayer anions such as carbonates.49 Hydrotalcites containing physically adsorbed water give strong water deformation modes at 1631 cm−1. This is attributed to adjacent water molecules in the hydrotalcite interlayers.48 The band at 1364 cm−1 was assigned to a carbonate bonded to the hydroxyl surface of the hydrotalcite.49 The band at 1024 cm−1 was assigned to the symmetric stretch of the free carbonate canions.48 The band at 777 cm−1 was assigned to the bending vibration of free carbonate anions. The IR absorption band at 443 cm−1 was assigned to Al–O bonds.50
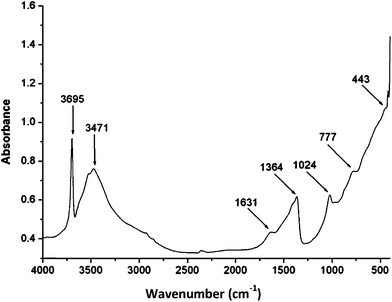 |
| | Fig. 1 FT-IR spectra of the hydrotalcite (Mg–Al–CO3). | |
Fig. 2 presents the XRD pattern of the hydrotalcites as well as the product of their thermal decomposition. The obtained results reveal that the prepared sample had a hexagonal structure with peaks characteristic of hydrotalcite. The Mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al atomic ratio was measured using X-ray microanalysis and was found to be 3.16, which is in good agreement with the metallic ratio (3.0) taken in solution. The value of x [x = MIII/MII + MIII] was found to be 0.24, which suggest the purity of the hydrotalcite.51 Thermal treatment of the sample resulted in the change of the chemical composition and phase content. It should be noticed that both the interlayer and weakly absorbed water were removed from the hydrotalcite sample at these temperatures (Fig. 2). The presence of a CO32− anion in the interlayer gallery of the hydrotalcite was confirmed by the characteristic basal spacing d003 = 7.76 Å and indicates a gallery height of 2.96 Å with the crystalline material.52 The ordering is achieved also in the stacking direction, as the (003), (006) and (009) diffraction lines become sharper and more intense. The crystallite size of this sample was found to be 24.87 nm as calculated using Scherrer's formula.53 More intensive and sharper reflections of the (003) and (006) planes was found at low 2θ values (11–23°). Fig. 2 shows a typical SEM image of the Mg–Al–CO3 hydrotalcite. As seen in this figure, the lamellar particles have a rounded hexagonal shape and are typical of a hydrotalcite-like material, which confirms the structure of the hydrotalcite; furthermore, the material was found to be mesoporous, with a surface area of 90 m2 g−1. Electron microscopy was chosen as a convenient tool to investigate the morphology of hydrotalcite-based materials and to investigate the effect of the preparation parameters on their morphology.
:Al atomic ratio was measured using X-ray microanalysis and was found to be 3.16, which is in good agreement with the metallic ratio (3.0) taken in solution. The value of x [x = MIII/MII + MIII] was found to be 0.24, which suggest the purity of the hydrotalcite.51 Thermal treatment of the sample resulted in the change of the chemical composition and phase content. It should be noticed that both the interlayer and weakly absorbed water were removed from the hydrotalcite sample at these temperatures (Fig. 2). The presence of a CO32− anion in the interlayer gallery of the hydrotalcite was confirmed by the characteristic basal spacing d003 = 7.76 Å and indicates a gallery height of 2.96 Å with the crystalline material.52 The ordering is achieved also in the stacking direction, as the (003), (006) and (009) diffraction lines become sharper and more intense. The crystallite size of this sample was found to be 24.87 nm as calculated using Scherrer's formula.53 More intensive and sharper reflections of the (003) and (006) planes was found at low 2θ values (11–23°). Fig. 2 shows a typical SEM image of the Mg–Al–CO3 hydrotalcite. As seen in this figure, the lamellar particles have a rounded hexagonal shape and are typical of a hydrotalcite-like material, which confirms the structure of the hydrotalcite; furthermore, the material was found to be mesoporous, with a surface area of 90 m2 g−1. Electron microscopy was chosen as a convenient tool to investigate the morphology of hydrotalcite-based materials and to investigate the effect of the preparation parameters on their morphology.
 |
| | Fig. 2 XRD pattern and SEM image of hydrotalcite (Mg–Al, 3:1). | |
The TG graph (Fig. 3) of the hydrotalcite (Al:Mg:CO3) exhibits a total mass of 38.26% at 330–570 °C, and other mass losses at 60–230 °C, 230–330 °C and 570–670 °C, respectively. The TG results indicate that the hydrotalcite (Al:Mg:CO3) is thermally stable up to 230 °C.54 The second mass loss between 223 and 330 °C was ascribed to the dehydroxylation of the brucite-like layers along with anion decomposition, leaving a Mg, Al oxo-hydroxide up to 330 °C. Finally, the third mass loss was assigned to progressive elimination of hydroxyl ions and the production of metal oxides and a spinel structure.
 |
| | Fig. 3 DTG curve of hydrotalcite (Mg–Al, 3:1). | |
The powder X-ray diffractogram of the Ca–Al–CO3 LDH sample given in Fig. 4 shows the structure of the hydrotalcite, displaying the characteristic reflection of (i) sharp and intense basal reflections of the 003 and 006 planes in the low angle, and (ii) intense reflections of the 009, 110 and 113 planes from the middle to the end angle. The LDH samples synthesized under different synthetic parameter have a similar structure; however, variations in the interlayer d-spacing of the characteristic 003 planes (d003), the crystallinity, and other structural features were observed to be affected by the synthesis parameters. The metallic ratios for Ca–Al–CO3 were observed by powder X-ray diffractometer and found to be 3:2:1. According to X-ray diffraction patterns, as shown in Fig. 4, the LDHs Ca–Al–CO3 sample have good systematic, narrow, sharp strong lines at low 2θ values (peaks close to 2θ = 11°, 24°, and 35°; ascribed to diffraction by the basal planes (003), (006) and (009), respectively) on the one hand, but broad and asymmetric ones at high 2θ values (peaks close to 2θ = 38°, 46°, and 60°; ascribed to diffraction by the (105), (108) and (110) planes,55 which are characteristic of clay minerals with a layered structure). Generally, the sharpness and intensity of the XRD peak is considered to be proportional to the crystallinity. A characteristic basal spacing d003 of 3.38 Å was confirmed in the interlayer gallery of the hydrotalcite. By XRD, the crystallite size of this sample was observed to be 47.025 nm. Fig. 4 shows the typical SEM image of Ca:Al LDH (3:1 molar ratio). As seen in this figure, the existing lamellar comprises almost homogeneous spherical aggregates with a hexagonal shape, which is the typical structure of hydrotalcite-like materials. However, big needle shaped particles are also shown. The LDH material shows high crystallinity, which shows a uniform distribution in the support.
 |
| | Fig. 4 P-XRD pattern and SEM image of hydrotalcite (Ca–Al, 3:1). | |
Chemistry
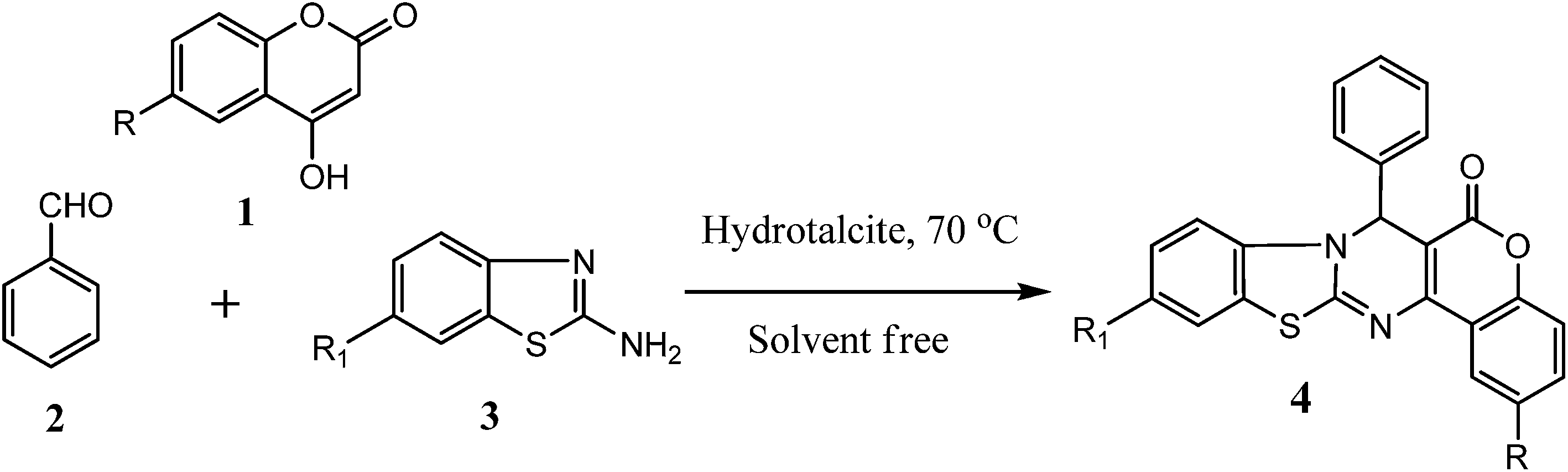
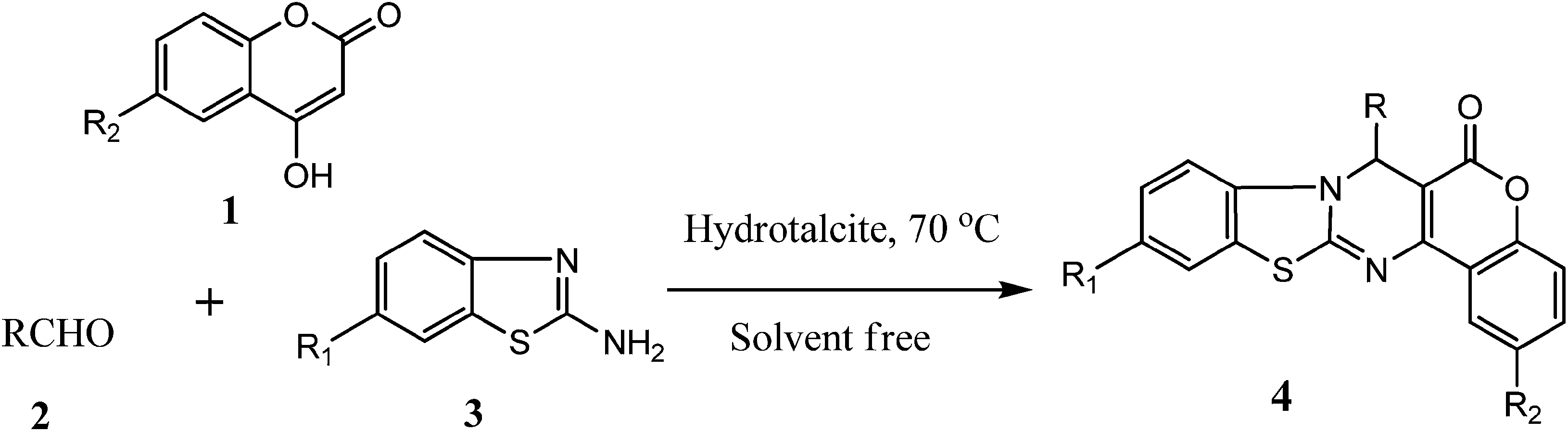
To optimize the reaction conditions, a model reaction of 4-hydroxy coumarin, benzaldehyde and 2-aminobenzothiazole was carried under solvent-free conditions. Under neat conditions, the reaction yield of the target product was too low, even when the reaction was performed at 90 and 120 °C for 10 h (Table 1, entries 2 and 3). Furthermore, to identify the best catalyst, different model reactions were performed with many catalysts, including hydrotalcites (Mg–Al–CO3 and Ca–Al–CO3) and different metal oxides, hydroxides and chlorides. The best result was obtained with Mg–Al hydrotalcite (Table 1, entry 4), while a slightly lower yield was obtained with Ca–Al hydrotalcite (Table 1, entry 5), and the other catalysts gave moderate yields of desired product (Table 1, entries 6–11). Mg–Al hydrotalcite catalyzes the reaction efficiently in a short reaction time and with a good yield. In order to evaluate the appropriate catalyst loading, a model reaction of benzaldehyde, 4-hydroxy coumarin and 2-aminobenzothiazole was carried out using 10 mg, 20 mg, 50 mg, 80 mg and 100 mg of hydrotalcite at 70 °C under solvent-free conditions (Table 1, entries 12–16). It was found that the reaction times and yields varied. On changing the amount of catalyst from 10–50 mg, the reaction time decreased and the yield increased. On the other hand, there was no significant improvement in reaction time and yield using a higher quantity of catalyst (100 mg). So, 80 mg of hydrotalcite as the catalyst was found to be the optimal quantity and was sufficient to catalyze the reaction at 70 °C under solvent-free conditions for the synthesis of chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one.
Table 1 Optimization of the reaction conditionsa
| Entry |
Catalyst (in gram) |
Time (h) |
Temperature (°C) |
Yieldb% of 4a |
| Reaction conditions: benzaldehyde (0.0025 mol), 4-hydroxy coumarin (0.0025 mol) and 2-amino benzothiazole (0.0025 mol), solvent-free conditions. Isolated yield. |
| 1 |
Neat |
10 |
70 |
15 |
| 2 |
Neat |
10 |
90 |
17 |
| 3 |
Neat |
10 |
120 |
16 |
| 4 |
Mg–Al–CO3, HT (0.08) |
2.0 |
70 |
95 |
| 5 |
Ca–Al–CO3, HT (0.08) |
2.0 |
70 |
86 |
| 6 |
AlCl3 (0.133) |
5.0 |
70 |
55 |
| 7 |
MgCl2 (0.095) |
4.0 |
70 |
62 |
| 8 |
Mg(OH)2 (0.058) |
4.0 |
70 |
41 |
| 9 |
Al(OH)3 (0.078) |
4.5 |
70 |
53 |
| 10 |
Ca(OH)2 (0.074) |
4.0 |
70 |
55 |
| 11 |
Al2O3 (0.101) |
4.5 |
70 |
60 |
| 12 |
Mg–Al–CO3, HT (0.01) |
6.0 |
70 |
78 |
| 13 |
Mg–Al–CO3, HT (0.02) |
4.0 |
70 |
85 |
| 14 |
Mg–Al–CO3, HT (0.05) |
3.0 |
70 |
89 |
| 15 |
Mg–Al–CO3, HT (0.08) |
2.0 |
70 |
95 |
| 16 |
Mg–Al–CO3, HT (0.10) |
2.0 |
70 |
95 |
The methodology involves the multi-component reaction of benzaldehyde (0.0025 mol), 4-hydroxy coumarine and 2-amino benzothiazole (0.0025 mol) using hydrotalcite (Mg–Al–CO3) as a catalyst and resulted in product formation within 2–3 h (Scheme 1). The structure of chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one was confirmed by 1H NMR, 13C NMR, mass spectra and elemental analyses. The NMR spectra of the product 4a was characterized by a singlet at 6.39 δ due to the C–H hydrogen and a multiplet at 7.05–7.17 δ, a triplet at 7.17 δ and a multiplet at 7.22–7.30 δ for three, two and four aromatic hydrogens, The hydrogen of the aromatic ring of benzothiazole exhibits a multiplet at 7.38–7.51 δ. The mass spectra of product 4a show a molecular ion peak at 383, supported by the good agreement with CHN elemental analysis. This supports the probable structure of product 4a. Other derivatives show the corresponding pattern, and the only difference is an additional peak that arises due to substitution at the aromatic ring of the aldehyde moiety in compounds such as, which demonstrated two additional peaks at 6.38 and 6.75 δ due to the two ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH and compound 4d, which showed a singlet at 3.08 δ for six hydrogens of the –N(CH3)2 group. A variety of electron-donating and electron-withdrawing groups on the aromatic aldehydes and benzothiazole have been studied (4a–4o) Table 2.
CH and compound 4d, which showed a singlet at 3.08 δ for six hydrogens of the –N(CH3)2 group. A variety of electron-donating and electron-withdrawing groups on the aromatic aldehydes and benzothiazole have been studied (4a–4o) Table 2.
 |
| | Scheme 1 Synthesis of chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one. | |
Table 2 Synthesis of chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one
Recyclability and reusability of hydrotalcite
For studying the recyclability and reusability, a model reaction was carried out using benzaldehyde, 4-hydroxy coumarin and 2-aminobenzothiazole by incorporating 80 mg of hydrotalcite as a catalyst (Fig. 5). After completion of the reaction, the contents were filtered to recycle the hydrotalcite catalyst. To remove any organic impurities, recycled hydrotalcite was washed with methanol. MgII hydrotalcite catalyst could be readily recovered and reused for at least five runs without any significant loss of activity (Table 3).
 |
| | Fig. 5 Recyclability and reusability of hydrotalcite catalyst. | |
Table 3 Reusability of hydrotalcite (Mg–Al–CO3, HT)
| Entry |
Number of runs |
Time (h) |
Yielda (%) |
| Isolated yield. |
| 1 |
Fresh HT |
2.0 |
95 |
| 2 |
HT (recycle I) |
2.0 |
94 |
| 3 |
HT (recycle II) |
2.5 |
94 |
| 4 |
HT (recycle III) |
2.5 |
93 |
| 5 |
HT (recycle IV) |
2.5 |
92 |
Leaching test
The filtrate was analyzed for leached metal content by ICP emission spectroscopy. No metal was detected. These results confirmed that metal leaching did not occur. Furthermore, the Mg–Al mixed oxide was found to be thermally and mechanically stable and no significant difference was observed in particle size and morphology of the used catalyst, as evidenced by SEM.56 To ensue the sustainability of the structure of the recovered hydrotalcite (Fig. 6), XRD analysis was carried out and showed its similar profile to the fresh catalyst, which confirmed that the layered structure of hydrotalcite was maintained after the reaction.
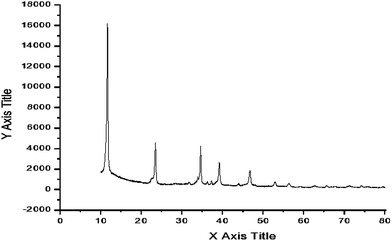 |
| | Fig. 6 XRD pattern of recovered hydrotalcite. | |
SEM analysis and effect of ageing time
To test if the ageing time could influence the crystal growth kinetically or thermodynamically, we considered ageing time as a parameter to control the particle size. To visualize the influence of ageing time on the crystallinity of the material, SEM images of the hydrotalcite samples at a Mg/Al molar ratio of 3.0 were recorded. The SEM images at different ageing times are shown in Fig. 7. A well-developed layered and platelet structure of the hydrotalcite is shown in the SEM image. The crystallinity of the hydrotalcite was observed to increase slowly up to 3 h at a temperature of 110 °C (the hydrothermal treatment temperature), followed by a sharp increase in the time range 6–9 h. On the other hand, poor crystallinity was observed at 0 h ageing time. The observations from the SEM images were in good agreement with the P-XRD pattern. These results confirmed an increase in the crystallinity of hydrotalcite sample at a Mg/Al ratio of 3.0 under hydrothermal treatment. From the results, it is clear that the crystallinity of the hydrotalcite sample depends on the ageing time and temperature conditions. The crystalline size of the hydrotalcite sample increased on increasing the ageing time. It seems that the ageing time has an effect on particle size, and it is thought that the particles may go through further crystal growth or intergrowth into larger particles (secondary particles) by the aggregation of primary particles upon ageing. High crystallinity prevented the formation of a poorly lamellar structure, and loss of the lamellar structure was the main cause of the catalyst deactivation.
 |
| | Fig. 7 SEM image of hydrotalcite with Mg/Al (3:1) at different ageing times at 110 °C. | |
Surface area measurement and XPS analysis
The surface area of the hydrotalcite samples was determined from the N2 adsorption data measured at 77 K. The samples were activated at 80 °C for 4 h under vacuum prior to the N2 adsorption measurements. The specific surface areas of the samples were calculated from the N2 adsorption isotherms according to the BET method, and are shown in Table 4. As seen in Table 4 and Fig. 9, as the metallic ratio of Mg/Al increases from 1:1 to 3:1, the surface area (Table 1) and basicity (Fig. 9) also increase.
Table 4 Specific surface area of synthesized hydrotalcite (Mg/Al)
| S. N. |
Mg/Al ratios |
SBET (m2 g−1) |
| 1 |
1:1 |
78 |
| 2 |
2:1 |
84 |
| 3 |
3:1 |
92 |
XPS measurements were performed using a Kratos AXIS HSi instrument equipped with a charge neutralizer and a Mg Kα X-ray source. Spectra were recorded at normal emission using an analyser pass energy of 20 eV and X-ray power of 144 W, and were energy referenced to the valence band and adventitious carbon. The surface composition of Mg–HT (before and after use) was studied by X-ray photoelectron spectroscopy (XPS) analysis (Fig. 8). The corresponding Mg 2p and Al 2s spectra are shown in Fig. 8(a) and (b). Both Fig. 8(a) and (b) exhibit a single broad feature indicative of a unique chemical environment. As the Mg2+ content rose across the series from Mg/Al (1:1) to Mg/Al (3:1), the binding energies of both features decreased. The Al 2 s BE (binding energy) for Mg/Al (3:1) was 119.1 eV, close to that of pure Al2O3 at 119.4 eV, and progressively shifted downwards to 118.5 eV for the Mg/Al (3:1) sample. The Mg 2p binding energy (BE) of the hydrotalcites likewise fell from 49.9 eV to 49.5 eV, approaching that of pure MgO at 49.0 eV. These changes reflect the change in intra-layer electron density in the ‘brucite-like’ layer as Mg2+ is replaced by Al3+. The O 1s for Al2O3 exhibited a single state at 530.9 eV binding energy (BE), characteristic of the hydroxyl environment.57 The O 1s binding energy changes with chemical composition, suggesting that the chemical form of the surface oxygen species depends on the chemical composition. Peak deconvolution and fitting revealed these comprise two well-defined components: the parent state at 530.9 eV, together with a new lower binding energy feature at ∼529.3 eV, characteristic of O2−.58 The increase in O2− character is consistent with the incorporation of Mg2+ centres into the hydrotalcite framework, and also in line with the Hammett basicity measurements shown in Fig. 9 and 10, which show that the strength of the strongest base sites of these materials increases with the Mg content.
 |
| | Fig. 8 (a) Mg 2p XP spectra, (b) Al 2s XP spectra for hydrotalcite materials and O 1s XP spectra. | |
 |
| | Fig. 9 Basicity of hydrotalcite with different metal ratios. | |
 |
| | Fig. 10 Basicity of 3.0 hydrotalcite at different temperatures. | |
The surface composition of the MgO, Al2O3 and Mg(1 − x)AlxO samples was measured by XPS. The XPS quantitative analysis results have been incorporated in Table 5. The binding energy (Table 5, column 2) changes with chemical composition, suggesting that the chemical form of the surface oxygen species depends on the chemical composition. The measured O 1s binding energy increases as the Al content increases. The amount of surface oxygen also depends on the chemical composition. As expected, the Als and Mgs values follow qualitative trends that parallel the bulk sample composition. As x increases, Als increased and Mgs decreased (Table 5). As the Al content in HT samples increased; however, the Als/Alb ratios decreased and Mgs/Mgb ratios increased. In HT samples with a low Al content, Mgs/Os ratios were very similar to those in MgO, but these ratios decreased as the Al content increased. The Als/Os ratios were between 0.3 and 0.6 in all the samples; these values are much lower than in pure Al2O3 (0.81).
Table 5 Surface composition of hydrotalcite by XPS analysis
| Samples |
Surface composition |
| O 1s B. E.a (eV) |
Mgs/Mgb |
Als/Alb |
Als/Os |
Mgs/Os |
| Binding energy. |
| MgO |
529.3 |
1.04 |
— |
— |
1.10 |
| Mg/Al (1:1) |
530.6 |
1.38 |
1.44 |
0.35 |
0.91 |
| Mg/Al (2:1) |
530.8 |
1.65 |
1.38 |
0.40 |
0.50 |
| Mg/Al (3:1) |
531.2 |
2.50 |
1.20 |
0.63 |
0.70 |
| Al2O3 |
530.9 |
— |
1.12 |
0.81 |
— |
Basicity of hydrotalcite
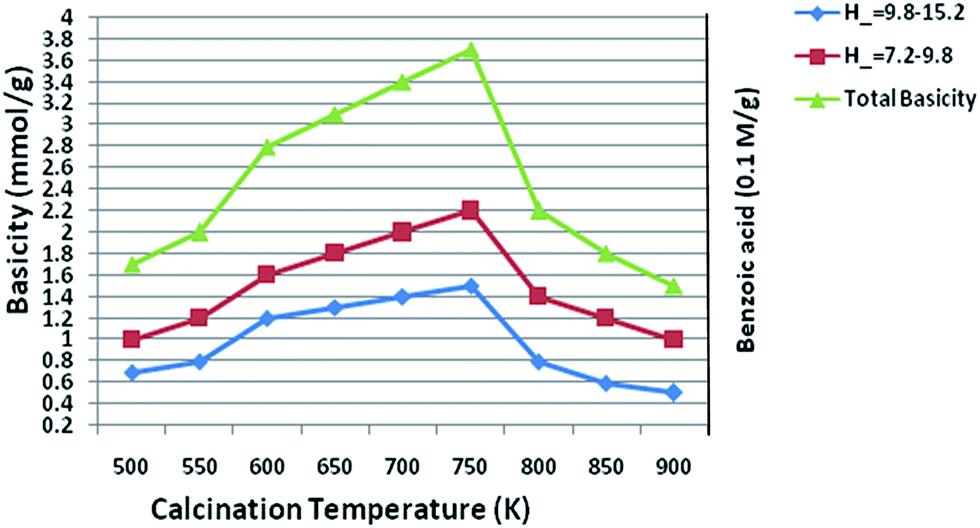
The basicity of the catalyst was determined using the Hammett indicator and benzoic acid titration method.59 From the results of Fig. 9, the basic strength of all the samples was found to be in the range of 9.3–15.0. The basicity results also revealed that calcined hydrotalcite possesses different types of surface basic sites, with the main basic sites H_ in the range of 7.2–9.8 and the other sites with H_ in the range of 9.8–15.0. Results were also reported by Di Cosimo et al.60 that suggested that calcined hydrotalcite contains outer surface basic sites of Mg/Al metal, while pure MgO possesses strong basic sites consisting predominantly of surface oxygen. The Hammett titration method was proven in the above study by our present experimental. Our results also revealed that the basic site responsible for the basicity of the hydrotalcite plays a vital role.
Furthermore, we found out from our study that basicity is related with the loading of the molar ratios of the Mg/Al metal contents. The results clearly demonstrate that the total basicity of the hydrotalcite (sample) increases gradually up to the optimum limit of the molar ratios of Mg/Al. Because the total basicity increases as the Mg/Al molar ratios reach up to the maximum value of 3.0, but the molar ratios increase from 3.0, it decreases further, with a resultant loss in the catalytic activity of the hydrotalcite. Our experimental studies were also proven quantitatively by other research, which have found similar trends as in the present study.61 Nakatsuka et al.'s61a and Fishel and Davis'61b groups also measured the number of basic sites or the basicity by titration with benzoic acid and by the TPD of CO2, respectively, and found similar results in that a maximum basic site density was observed at a Mg/Al ratio of 2.6 and 3.0, respectively. So, a molar ratio of Mg/Al of 3.0 was found to be optimum for the best results and, therefore, we measured the basicity of 3.0 HT calcined at different temperatures. From our study or data (Fig. 10), it was clearly proven that a 750 K calcinations temperature was optimum because of the maximum basicity (reaching 3.6 mmol g−1) being reached at this temperature. Below 573 K and above 750 K temperature, the basicity level was too low. In terms of the catalytic activity of hydrotalcite, basicity plays a major role and it could be expected that the catalytic activity increases with an increase in the basicity.
A plausible mechanism for the synthesis of chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one derivatives (4) is given in Scheme 2. In the beginning, the mechanism involves Knoevenagel condensation of the 4-hydroxycoumarin (1) and aldehyde (2) in the presence of hydrotalcite, producing an intermediate, whereby the basic sites of the catalyst abstract an acidic proton of malononitrile and then a subsequent attack on the carbonyl group furnishes the condensed product I.62 This is followed by the Michael addition of 2-aminobenzothiazole (3) to the CC bond of intermediate (A) to form intermediate (B) through tautomerization. Then, an intramolecular cyclic condensation between the amino and the carbonyl groups of the Michael adduct B occurs to afford intermediate (C), which then affords the desired compounds (4) on dehydration.
 |
| | Scheme 2 A plausible reaction mechanism. | |
Materials and methods
Experimental
The 1H NMR spectra were measured using a BRUKER AVANCE II 400 NMR spectrometer with tetramethylsilane as an internal standard at 20–25 °C; data for 1H NMR are reported as follows: chemical shift (ppm), integration, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet and br, broad), coupling constant (Hz). IR spectra were recorded by a SHIMADZU IR spectrometer for a sample dispersed in a KBr pellet or Nujol, reported in terms of the frequency of absorption (cm−1). E-Merck pre-coated TLC plates and RANKEM silica gel G were used for the preparative thin-layer chromatography. All melting points were determined in open capillaries and are uncorrected. AR grade 4-hydroxy coumarin, aldehydes and other catalysts were purchased from Himedia Laboratory Ltd., Mumbai, India. 2-Amino benzothiazole was purchased from Sigma Aldrich and used without further purification.
Preparation of hydrotalcite (HT)
Typical procedure. Al2O3 (1.02 g) was suspended in distilled water (2 ml) and magnesium hydroxide (3.5 g) was added to the mixture, and the contents were stirred giving a pH of 8. Then, sodium bicarbonate (2.4 g) was added to bring the pH of the mixture to 10. The mixture was ground with a mortar and pestle for 5 min at room temperature, and the resulting white product was filtered and repeatedly washed with distilled water and dried at 100 °C.
Typical procedure for the synthesis of fused pyrimidines (4a–4o). A mixture of aldehydes (0.0025 mol), 4-hydroxycoumarin (0.0025 mol) and 2-amino benzothiazole (0.0025 mol) were heated at 70 °C under solvent-free conditions using hydrotalcite as a catalyst. After completion of the reaction (by TLC analysis), the reaction mixture was cooled to room temperature and poured into cold water. Then, the solid mass obtained was dissolved in ethanol and filtered. The solid hydrotalcite was separated out as a solid. The product was recrystallized in ethanol. Hydrotalcite was washed with ethanol to remove organic impurities.
Reusability of the hydrotalcite
Recycled hydrotalcite was reused for five times as such with a mixture of aldehydes (0.0025 mol), 4-hydroxycoumarin (0.0025 mol) and 2-amino benzothiazole (0.0025 mol) at 70 °C. The yield and time taken in each of the five runs are given in Table 3.
Characterization data
7-Phenylchromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4a). White powder, mp 200–202 °C; 1H NMR (500 MHz, DMSO d6): δH 6.23 (s, 1H, –CH), 7.05–7.17 (m, 3H, Ar-H), 7.17 (t, 2H, J = 8.0 Hz, Ar-H), 7.22–7.30 (m, 4H, Ar-H), 7.38–7.51 (m, 4H); 13C NMR (125 MHz, DMSO d6): 68, 103, 114, 115, 115, 119, 122, 122, 123, 123, 124, 126, 126, 127, 127, 131, 141, 152, 164, 167, 168; ESI-MS: m/z calculated for C23H14N2O2S 382.43 found [M + H]+ 383.
7-Styrylchromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4b). Light brown crystal, mp 180–182; 1H NMR (500 MHz, DMSO d6): δH 5.63 (s, 1H, –CH), 6.26 (d, 1H, CH), 6.70 (d, 1H, CH), 7.19–7.38 (m, 8H, Ar-H), 7.58 (t, 2H, J = 7.5 Hz, Ar-H), 7.84 (t, 3H, J = 8.0 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 65, 101, 102, 103, 112, 114, 116, 117, 120, 122, 124, 128, 129, 131, 133, 137, 144, 151, 152, 157, 161, 163, 164, 167; ESI-MS: m/z calculated for C25H16N2O2S 408.47 found [M + H]+ 409.
7-(4-Chlorophenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4c). Light yellow powder, mp 188–189; 1H NMR (500 MHz, DMSO d6): δH 6.39 (s, 1H, –CH), 6.94–7.09 (m, 2H, Ar-H), 7.16 (d, 1H, J = 8.0 Hz, Ar-H), 7.21 (t, 1H, J = 7.5 Hz, Ar-H), 7.39–7.62 (m, 4H, J = 8.0 Hz), 7.82 (d, 2H, J = 8.5 Hz, Ar-H), 7.90 (d, 2H, J = 7.0 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 68, 106, 113, 118, 118, 121, 125, 125, 125, 126, 129, 130, 134, 146, 155, 163, 167, 170; ESI-MS: m/z calculated for C23H13ClN2O2S 416.88 found [M + H]+ 418.
7-(4-Dimethylaminophenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4d). Reddish brown powder, mp 160–162; 1H NMR (500 MHz, DMSO d6): δH 3.08 (s, 6H, –N(CH3)2), 6.26 (s, 1H, –CH), 7.15–7.41 (m, 8H, Ar-H), 7.59 (t, 2H, J = 7.5 Hz, Ar-H), 7.86 (d, 2H, J = 7.5 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 45, 65, 103, 111, 114, 115, 118, 118, 119, 121, 123, 124, 125, 126, 128, 131, 131, 141, 152, 164, 167; ESI-MS: m/z calculated for C25H19N3O2S 425.50 found [M + H]+ 426.
7-(3-Hydroxyphenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4e). Off white powder, mp 200–202 °C; 1H NMR (500 MHz, DMSO d6): δH 6.20 (s, 1H, –CH), 6.45–6.56 (m, 3H, Ar-H), 6.94 (t, 1H, J = 7.5 Hz, Ar-H), 7.22–7.28 (m, 4H, Ar-H), 7.41–7.51 (m, 4H, Ar-H), 9.26 (br, 1H, OH); 13C NMR (125 MHz, DMSO d6): 65, 103, 112, 113, 114, 115, 117, 119, 122, 122, 123, 123, 124, 127, 128, 131, 141, 143, 152, 157, 164, 167, 168; ESI-MS: m/z calculated for C23H14N2O3S 398.43 found [M + H]+ 399.
7-(4-Methoxyphenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4f). Off white powder, mp 234–236 °C; 1H NMR (500 MHz, DMSO d6): δH 3.73 (s, 3H, OCH3), 6.24 (s, 1H, –CH), 6.80 (d, 2H, J = 7.0 Hz, Ar-H), 7.16–7.43 (m, 4H, Ar-H), 7.51 (t, 2H, J = 7.5 Hz, Ar-H), 7.68 (d, 2H, J = 7.0 Hz, Ar-H), 7.79 (d, 2H, J = 7.5 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 56, 66, 103, 118, 121, 125, 128, 133, 133, 133, 134, 138, 141, 142, 145, 150, 152, 164, 166; ESI-MS: m/z calculated for C24H16N2O3S 412.46 found [M + H]+ 413.
7-(4-Methylphenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4g). Yellow powder, mp > 250 °C; 1H NMR (500 MHz, DMSO d6): δH 2.80 (s, 3H, CH3), 6.24 (s, 1H, –CH), 7.19–7.40 (m, 8H, Ar–H), 7.51 (t, 2H, J = 7.5 Hz, Ar-H), 7.80 (d, 2H, J = 7.0 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 27, 65, 102, 111, 116, 119, 119, 123, 123, 124, 124, 128, 131, 140, 145, 152, 154, 164, 167; ESI-MS: m/z calculated for C24H16N2O2S 396.46 found [M]+ 397.
7-(4-Nitrophenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4h). Off white powder, mp 200–202 °C; 1H NMR (500 MHz, DMSO d6): δH 6.17 (s, 1H, –CH), 7.20–7.25 (m, 5H, Ar-H), 7.29 (d, 2H, J = 7.0 Hz, Ar-H), 7.50 (t, 2H, J = 7.5 Hz, Ar-H), 7.81 (d, 2H, J = 7.5 Hz, Ar-H), 8.13 (d, 2H, J = 7.5 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 67, 103, 111, 115, 118, 122, 123, 123, 124, 124, 126, 128, 128, 131, 144, 152, 161, 164, 167; ESI-MS: m/z calculated for C23H13N3O4S 427.43 found [M + H]+ 429.
7-(2-Methylphenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4i). Off white powder, mp > 250 °C; 1H NMR (500 MHz, DMSO d6): δH 2.61 (s, 3H, CH3), 6.37 (s, 1H, –CH), 7.21–7.25 (m, 4H, Ar–H), 7.29 (t, 2H, J = 7.0 Hz, Ar-H), 7.46 (t, 2H, J = 7.5 Hz, Ar-H), 7.86 (d, 2H, J = 7.5 Hz, Ar-H), 8.13 (d, 2H, J = 7.5 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 38, 67, 103, 115, 118, 123, 123, 124, 124, 128, 128, 130, 131, 140, 144, 150, 152, 164, 166; ESI-MS: m/z calculated for C24H16N2O2S 396.46 found [M + H]+ 397.
7-(2-Methoxyphenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4j). Off white powder, mp 234–236 °C; 1H NMR (500 MHz, DMSO d6): δH 3.73 (s, 3H, OCH3), 6.24 (s, 1H, –CH), 6.78 (d, 2H, J = 7.0 Hz, Ar-H), 7.19–7.31 (m, 4H, Ar-H), 7.53 (t, 2H, J = 7.5 Hz, Ar-H), 7.71 (d, 2H, J = 7.0 Hz, Ar-H), 7.79 (d, 2H, J = 7.5 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 55, 66, 106, 115, 119, 122, 122, 123, 123, 126, 131, 134, 135, 143, 149, 152, 156, 167, 169; ESI-MS: m/z calculated for C24H16N2O3S 412.46 found [M + H]+ 413.
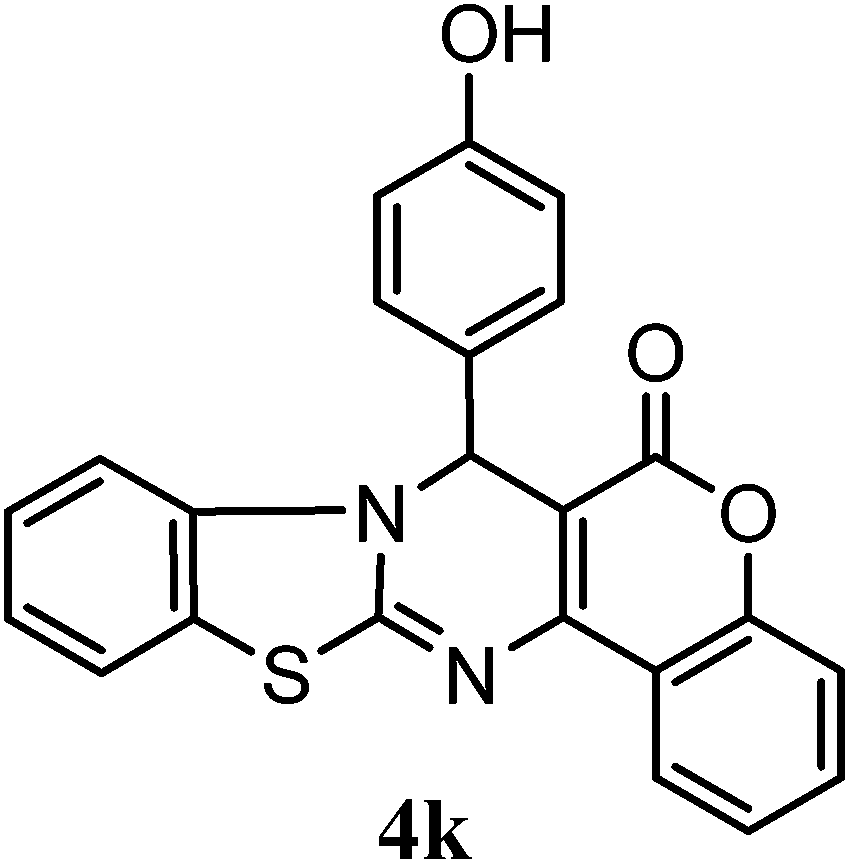
7-(4-Hydroxyphenyl)chromeno[4,3-d]benzothiazolo[3,2-a]pyrimidin-6(7H)-one (4k). Off white powder, mp 200–202 °C; 1H NMR (500 MHz, DMSO d6): δH 6.16 (s, 1H, –CH), 6.54 (d, 1H, J = 7.5 Hz, Ar-H), 6.86 (d, 1H, J = 7.5 Hz, Ar-H), 7.19–7.26 (m, 4H, Ar-H), 7.35–7.51 (m, 4H, Ar-H), 7.80 (d, 2H, J = 7.5 Hz, Ar-H), 8.91 (br, 1H, OH); 13C NMR (125 MHz, DMSO d6): 65, 101, 109, 111, 112, 113, 115, 117, 120, 120, 120, 121, 124, 125, 128, 138, 141, 149, 159, 154, 162, 164, 166, 167; ESI-MS: m/z calculated for C23H14N2O3S 398.43 found [M + H]+ 399.
7-(4-Chloroyphenyl)-11-methylbenzo[4,5]thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6(7H)-one (4l). Off white powder, mp 184–186 °C; 1H NMR (500 MHz, DMSO d6): δH 2.29 (s, 3H, CH3), 6.29 (s, 1H, –CH), 7.14 (d, 2H, J = 7.5 Hz, Ar-H), 7.24–7.35 (m, 4H, Ar-H), 7.55–7.59 (m, 2H, Ar-H), 7.88 (d, 2H, J = 7.5 Hz, Ar-H), 7.93 (d, 1H, J = 8.5 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 23, 67, 104, 116, 116, 117, 117, 123, 128, 129, 129, 129, 130, 131, 132, 138, 139, 152, 164, 166; ESI-MS: m/z calculated for C24H15ClN2O2S 430.91 found [M + H]+ 431.
11-Nitro-7-phenyl-11-methylbenzo[4,5]thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6(7H)-one (4m). Off white powder, mp 215–217 °C; 1H NMR (500 MHz, DMSO d6): δH 6.37 (s, 1H, –CH), 7.27 (t, 2H, J = 8.0 Hz, Ar-H), 7.33 (d, 2H, J = 8.5 Hz, Ar-H), 7.40 (d, 2H, J = 8.5 Hz, Ar-H), 7.56 (t, 2H, J = 8.0 Hz, Ar-H), 7.84 (d, 2H, J = 8.0 Hz, Ar-H), 8.09 (d, 2H, J = 9.0 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 69, 103, 111, 115, 118, 122, 122, 123, 127, 130, 131, 139, 145, 149, 151, 157, 164, 166; ESI-MS: m/z calculated for C23H13N3O4S 427.43 found [M]+ 427.5.
2-Chloro-7-(p-tolyl)benzo[4,5]thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6(7H)-one (4n). Off white powder, mp 225–227 °C; 1H NMR (500 MHz, DMSO d6): δH 2.39 (s, 3H, CH3), 6.30 (s, 1H, –CH), 6.89–6.94 (dd, 2H, J = 8.0, 8.5 Hz, Ar-H), 7.06 (t, 2H, J = 8.0 Hz), 7.25–7.36 (m, 4H, Ar-H), 7.70 (d, 1H, J = 7.5 Hz, Ar-H), 8.10 (d, 2H, J = 8.5 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 21, 69, 102, 117, 118, 121, 121, 121, 126, 127, 128, 129, 129, 129, 130, 1132, 134, 143, 150, 151, 161, 163, 167; ESI-MS: m/z calculated for C24H15ClN2O2S 430.90 found [M]+ 430.9.
7-(4-Bromophenyl)-2-chloro-11-methylbenzo[4,5]thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6(7H)-one (4o). Off white powder, mp > 250 °C; 1H NMR (500 MHz, DMSO d6): δH 2.23 (s, 3H, CH3), 6.29 (s, 1H, –CH), 7.09–7.23 (m, 8H, Ar-H), 7.32 (t, 2H, J = 7.5 Hz, Ar-H); 13C NMR (125 MHz, DMSO d6): 22, 66, 104, 107, 113, 117, 125, 126, 128, 129, 130, 130, 134, 143, 151, 155, 162, 164; ESI-MS: m/z calculated for C24H14BrClN2O2S 509.80 found [M]+ 509.8.
Acknowledgements
We are grateful thanks to Chandigarh and Punjab University, Chandigarh for spectral analytical data.
References
- C. Oliver Kappe, Tetrahedron, 1993, 49, 6937 CrossRef.
- K. S. Atwal, G. C. Rovnyak, B. C. O”Reilly and J. Schwartz, J. Org. Chem., 1989, 54, 5898 CrossRef CAS.
-
(a) K. S. Atwal, B. N. Swanson, S. E. Unger, D. M. Floyd, S. Moreland, A. Hedberg and B. C. O’reilly, J. Med. Chem., 1991, 34, 806 CrossRef CAS PubMed;
(b) G. C. Rovnyak, K. S. Atwal, A. Hedberg, S. D. Kimball, S. Moreland, J. Z. Gougoutas, B. C. O'Reillly, J. Schwartz and M. F. Malley, J. Med. Chem., 1992, 35, 3254 CrossRef CAS PubMed.
-
(a) G. C. Rovnyak, S. D. Kimbal, B. Beyer, G. Cucinotta, J. D. Dimarco, J. Gougoutas, A. Hedberg, M. Malley, J. P. MaCaethy, R. Zhang and S. Mereland, J. Med. Chem., 1995, 38, 119 CrossRef CAS;
(b) C. O. Kappe, W. M. F. Fabian and M. A. Semons, Tetrahedron, 1997, 53, 2803 CrossRef CAS.
- K. R. Lanjewar, A. M. Rahatgaonkar, M. S. Chorghade and B. D. Saraf, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2009, 48, 1732 Search PubMed.
-
(a) M. M. Heravi, L. Ranjwar, F. Deriknand, B. Alimadadi and H. A. Oskooie, Mol. Diversity, 2008, 12, 181 CrossRef CAS PubMed;
(b) S. Chang, J. S. Ji and L. Yu, J. Chin. Chem. Soc., 2008, 55, 292 CrossRef;
(c) N. K. Shah, M. P. Patel and R. G. Patel, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 2704 CrossRef CAS.
- R. Kumar, S. Malik and R. Chamdra, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2009, 48, 718 Search PubMed.
- J. Kempson, W. J. Pitts, J. Barbosa, J. Guo, O. Omotoso, A. Watson, K. Stebbins, G. C. Starling, J. H. Dodd, J. C. Barrish, R. Felix and K. Fischer, Bioorg. Med. Chem. Lett., 2005, 15, 1829 CrossRef CAS PubMed.
- O. Bruno, C. Brullo, S. Schenone, F. Bondavalli, A. Ranise, M. Tognolini, M. Impicciatore, V. Ballabeni and E. Barocelli, Bioorg. Med. Chem., 2006, 14, 121 CrossRef CAS PubMed.
- S. A. Said, A. E.-G. Amr, N. M. Sabry and M. M. Abdalla, Eur. J. Med. Chem., 2009, 44, 4787 CrossRef CAS PubMed.
- M. M. A. El-Gendy, M. Shaaban, K. A. Shaaban, A. M. El-Bondkly and H. Laatsch, J. Antibiot., 2008, 61, 149 CrossRef CAS PubMed.
- R. Magan, C. M. J. M. Salas, M. Barrera-Perez, M. J. Rosales and M. Sanchez-Moreno, Mem. Inst. Oswaldo Cruz, 2004, 99, 651 CrossRef CAS PubMed.
- I. Lakomska, Inorg. Chim. Acta, 2009, 362, 669 CrossRef CAS.
- O. I. El-Sabbagh, M. M. Baraka, S. M. Ibrahim, C. Pannecouque, G. Andrei, R. Snoeck, J. Balzarini and A. A. Rashad, Eur. J. Med. Chem., 2009, 44, 3746 CrossRef CAS PubMed.
-
(a) V. Zaharia, A. Ignat, N. Palibroda, B. Ngameni, V. Kuete, C. N. Fokunang, M. L. Moungang and B. T. Ngadjui, Eur. J. Med. Chem., 2010, 45, 5080 CrossRef CAS PubMed;
(b) I. Hutchinson, S. A. Jennings, B. R. Vishnuvajjala, A. D. Westwell and M. F. G. Stevens, J. Med. Chem., 2002, 45, 744 CrossRef CAS PubMed.
- A. Kamal, M. N. A. Khan, K. S. Reddy and K. Rohini, Bioorg. Med. Chem., 2007, 15, 1004 CrossRef CAS PubMed.
- R. B. Pathak, B. Jahan and S. C. Bahel, Bokin Bobai, 1981, 9, 477 CAS.
- D. D. Erol, U. Caliş, R. Demirdamar, N. Yulug and M. Ertan, J. Pharm. Sci., 1995, 84, 462 CrossRef CAS PubMed.
- F. Azam, B. A. El-Gnidi, I. A. Alkskas and M. A. Ahmed, J. Enzyme Inhib. Med. Chem., 2010, 25, 818 CrossRef CAS PubMed.
- Y. Kumar, R. Green, K. Z. Boryska, D. D. Wise, L. L. Wotring and L. B. Townsend, J. Med. Chem., 1993, 36, 3843 CrossRef CAS PubMed.
- R. N. Sharma, F. P. Xavier, K. K. Vasu, S. C. Chaturvedi and S. S. Pancholi, J. Enzyme Inhib. Med. Chem., 2009, 24, 890 CrossRef CAS PubMed.
-
(a) R. D. H. Murray, J. Mendez and S. A. Brown, Chemistry and Biochemistry, John Wiley & Sons Ltd, New York, 1982, p. 21 Search PubMed;
(b) M. M. Garazd, Y. L. Garadz and V. P. Khilya, Chem. Nat. Compd., 2003, 39, 54 CrossRef CAS.
-
(a) J. R. Hwu, R. Singha, S. C. Hong, Y. H. Chang, A. R. Das, I. Vliegen, E. D. Clercq and J. J. Neyts, Antiviral Res., 2008, 77, 157 CrossRef CAS PubMed;
(b) J. Neyts, E. D. Clercq, R. Singha, Y. H. Chang, A. R. Das, S. K. Chakraborty, S. C. Hong, S. C. Tsay, M. H. Hsu and J. R. Hwu, J. Med. Chem., 2009, 52, 1486 CrossRef CAS PubMed;
(c) J. R. Hwu, S. Y. Lin, S. C. Tsay, E. D. Clercq, P. Leyssen and J. Neyts, J. Med. Chem., 2011, 54, 2114 CrossRef CAS PubMed.
-
(a) Z. P. Li, J. F. Hu, M. N. Sun, H. J. Ji, S. F. Chu, G. Liu and N. H. Chen, Int. Immunopharmacol., 2012, 14, 145 CrossRef CAS PubMed;
(b) Z. P. Li, J. F. Hu, M. N. Sun, H. J. Ji, M. Zhao, D. H. Wu, G. Y. Li, G. Liu and N. H. Chen, Eur. J. Pharmacol., 2011, 661, 118 CrossRef CAS PubMed.
- Y. Shi and C. H. Zhou, Bioorg. Med. Chem. Lett., 2011, 21, 956 CrossRef CAS PubMed.
-
(a) F. Meggio, M. A. Pagano, S. Moro, G. Zagotto, M. Ruzzene, S. Sarno, G. Cozza, J. Bain, M. Elliott, A. D. Deana, A. M. Brunati and L. A. Pinna, Biochemistry, 2004, 43, 12931 CrossRef CAS PubMed;
(b) A. Chilin, R. Battistutta, A. Bortolato, G. Cozza, S. Zanatta, G. Poletto, M. Mazzorana, G. Zagotto, E. Uriarte, A. Guiotto, L. A. Pinna, F. Meggio and S. Moro, J. Med. Chem., 2008, 51, 752 CrossRef CAS PubMed;
(c) G. L. Bras, C. Radanyi, J. F. Peyrat, J. D. Brion, M. Alami, V. Marsaud, B. Stella and J. M. Renoir, J. Med. Chem., 2007, 50, 6189 CrossRef PubMed;
(d) H. P. Zhao, A. C. Donnelly, B. R. Kusuma, G. E. L. Brandt, D. Brown, R. A. Rajewski, G. Vielhauer, J. Holzbeierlein, M. S. Cohen and B. S. J. Blagg, J. Med. Chem., 2011, 54, 3839 CrossRef CAS PubMed;
(e) H. P. Zhao, B. Yan, L. B. Peterson and B. S. J. Blagg, ACS Med. Chem. Lett., 2012, 3, 327 CrossRef CAS PubMed;
(f) A. C. Donnelly, J. R. Mays, J. A. Burlison, J. T. Nelson, G. Vielhauer, J. Holzbeierlein and B. S. J. Blagg, J. Org. Chem., 2008, 73, 8901 CrossRef CAS PubMed;
(g) A. Purohit, L. W. L. Woo, B. V. L. Potter and M. J. Reed, Cancer Res., 2000, 60, 3394 CAS;
(h) L. W. L. Woo, A. Purohit, B. Malini, M. J. Reed and B. V. L. Potter, Chem. Biol., 2000, 7, 773 CrossRef CAS PubMed;
(i) B. Malini, A. Purohit, D. Ganeshapillai, L. W. L. Woo, B. V. L. Potter and M. J. Reed, J. Biochem. Mol. Biol., 2000, 75, 253 CAS.
-
(a) M. Khaldi, A. D. Roy, M. Chaouch and J. P. Besse, J. Solid State Chem., 1997, 130, 66 CrossRef CAS;
(b) N. Gutmann and B. Muller, J. Solid State Chem., 1996, 122, 214 CrossRef CAS;
(c) F. Kooli, I. Crespo, C. Barriya, M. A. Ulibrarri and V. Rives, J. Mater. Chem., 1996, 6, 1199 RSC.
- N. Ahmet, Z. K. Birgul and M. Z. Luis, Z. Anorg. Allg. Chem., 2009, 635, 1470 CrossRef.
-
(a) M. S. Newton, J. D. Morrison, R. D. Pennell, N. P. Phodes and A. J. Toft, US Pat. Appl. 203152, 2010;
(b) V. Rives, Layered double hydroxides: present and future, Nova Science Publishers, New York, 2001 Search PubMed;
(c) S. K. Sharma, P. A. Parikh and R. V. Jarra, J. Mol. Catal. A: Chem., 2007, 278, 135 CrossRef CAS;
(d) C. O. Veloso, C. N. Perez, B. M. de Souza, E. C. Lima, A. G. Dias, J. L. F. Monteiro and C. A. Henriques, Microporous Mesoporous Mater., 2008, 107, 23 CrossRef CAS.
-
(a) W. Kagunya, Z. Hassan and W. Janes, Inorg. Chem., 1994, 35, 5970 CrossRef;
(b) M. Ulibarri, F. M. Labajos, V. Rives, R. Trujillano, W. Kagunya and W. Janes, Inorg. Chem., 1994, 33, 2592 CrossRef CAS.
- C. M. Jinesh, C. A. Antonyraj and S. Kannan, Catal. Today, 2009, 141, 176 CrossRef CAS.
-
(a) M. A Ulibarri, I. Pavlovic, C. Barriga, M. C. Hermosin and J. Cornejo, Appl. Clay Sci., 2001, 18, 17 CrossRef;
(b) G. Centi and S. Perathoner, Microporous Mesoporous Mater., 2008, 107, 3 CrossRef CAS.
- K. Motocura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani, K. Htsukawa and K. Kanedar, Chem.–Eur. J., 2006, 12, 8228 CrossRef PubMed.
- M. Turco, G. Bagnasco, U. Costantino, F. Marmottini, T. Montanari, G. Ramis and G. Busca, J. Catal., 2004, 228, 56 CAS.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, Catal. Rev.: Sci. Eng., 2001, 43, 443 CAS.
- Y. Xi and R. J. Davis, J. Catal., 2008, 254, 190 CrossRef CAS.
- S. K. Yun and T. Pinnavaia, J. Chem. Mater., 1995, 7, 348 CrossRef CAS.
- H. P. Boehm, J. Steinle and C. Vieweger, Angew. Chem., Int. Ed. Engl., 1977, 16, 265 CrossRef.
-
(a) A. De Roy, C. Forano, K. El Malki and J. P. Besse, in Expanded Clays and Other Microporous Solids, ed. M. L. Occelli and H. E. Robson, Reinhold, New York, 1992, vol. 2, p. 108 Search PubMed;
(b) F. Kovanda, T. Grygar and V. Dornicak, Solid State Sci., 2003, 5, 1019 CrossRef CAS;
(c) J. J. Creasey, A. Chieregato, J. C. Manayil, C. M. A. Parlett, K. Wilson and A. F. Lee, Catal. Sci. Technol., 2014, 4, 861 RSC.
- R. M. Taylor, Clay Miner., 1984, 19, 591 CAS.
- M. A. Dredzon, Inorg. Chem., 1988, 27, 4628 CrossRef.
-
(a) H. Nijs, A. Clearfield and E. F. Vansant, Microporous Mesoporous Mater., 1998, 23, 97 CrossRef CAS;
(b) H. Tamura, J. Chiba, M. Ito, T. Takeda, S. Kikkawa, Y. Mawatari and M. Tabata, J. Colloid Interface Sci., 2006, 300, 648 CrossRef CAS PubMed.
- S. P. Paredes, G. Fetter, P. Bosch and S. Bulbulian, J. Mater. Sci., 2006, 41, 3377 CrossRef CAS.
- F. M. Labajos, V. Rives and M. A. Ulibarri, J. Mater. Sci., 1992, 27, 1546 CrossRef CAS.
- S. Miyata, Clays Clay Miner., 1980, 282, 305 Search PubMed.
- O. Clause, M. Gazzano, F. Trifiro, A. Vaccari and L. Zatorski, Appl. Catal., 1991, 73, 217 CrossRef CAS.
-
(a) P. K. Sahu, P. K. Sahu, S. K. Gupta, D. Thavaselvam and D. D. Agarwal, Eur. J. Med. Chem., 2012, 54, 366 CrossRef CAS PubMed;
(b) P. K. Sahu, P. K. Sahu, D. Thavaselvam, A. M. Alafeefy and D. D. Agarwal, Med. Chem. Res., 2015, 24, 725 CrossRef CAS;
(c) P. K. Sahu, P. K. Sahu and D. D. Agarwal, RSC Adv., 2013, 3, 9854 RSC;
(d) P. K. Sahu, P. K. Sahu, S. K. Gupta and D. D. Agarwal, Ind. Eng. Chem. Res., 2014, 53, 2085 CrossRef CAS;
(e) P. K. Sahu, P. K. Sahu and D. D. Agarwal, RSC Adv, 2014, 4, 40414 RSC;
(f) P. K. Sahu, P. K. Sahu, Y. Sharma and D. D. Agarwal, J. Heterocycl. Chem., 2014, 51, 1193 CrossRef CAS;
(g) P. K. Sahu, P. K. Sahu and D. D. Agarwal, J. Indian Chem. Soc., 2015, 92, 169 CAS.
- S. J. Palmer, T. Nauyen and R. L. Frost, Coord. Chem. Rev., 2009, 253, 250 CrossRef CAS.
- S. J. Palmer and R. L. Frost, Ind. Eng. Chem. Res., 2011, 50, 5346 CrossRef CAS.
- M. Valcheva-Traykova, N. Davidova and A. Weiss, J. Mater. Sci., 1993, 28, 2157 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- F. M. Labojos, V. Rives and M. A. Ulibarri, J. Mater. Sci., 1992, 27, 1546 CrossRef.
- A. S. Bookin and A. Dritis, Clays Clay Miner., 1993, 41, 551 CAS.
- W. T. Reichle, Chem. Technol., 1986, 16, 58 CAS.
- K. R. Kottapalli, G. Monique, S. V. Jaime and F. Francois, J. Catal., 1998, 173, 115 CrossRef.
-
(a) Y. Liu, E. Lotero, J. J. Goodwin and X. Mo, Appl. Catal., A, 2007, 331, 138 CrossRef CAS;
(b) P. Chauyplod and W. Trakarnpruk, Ind. Eng. Chem. Res., 2009, 48, 4177 CrossRef.
- T. L. Barr, S. Seal, K. Wozniak and J. Klinowski, J. Chem. Soc., Faraday Trans., 1997, 93, 181 RSC.
- X. D. Peng, D. A. Richards and P. C. Stair, J. Catal., 1990, 121, 99 CrossRef CAS.
-
(a) D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183 CrossRef CAS;
(b) J. M. Fraile, N. Garcia, J. A. Mayoral, E. Pires and L. Rodan, Appl. Catal., A, 2009, 364, 87 CrossRef CAS;
(c) H. J. Hattori, J. Jpn. Pet. Inst., 2004, 47, 67 CrossRef CAS.
- J. J. Di Cosimo, V. K. Di’ez, M. Xu, E. Iglesia and C. R. Apesteguia, J. Catal., 1998, 178, 499 CrossRef CAS.
-
(a) T. Nakatsuka, H. Kawasaki, S. Yamashita and S. Kohjiya, Bull. Chem. Soc. Jpn., 1979, 52, 2449 CrossRef CAS;
(b) C. T. Fishel and R. Davis, Langmuir, 1994, 10, 159 CrossRef CAS.
-
(a) R. Maggi, R. Ballini, G. Sartori and R. Sartorio, Tetrahedron Lett., 2004, 45, 2297 CrossRef CAS;
(b) Y. M. Ren and C. Cai, Catal. Commun., 2008, 9, 1017 CrossRef CAS;
(c) S. Abdolmohammadi and S. Balalaie, Tetrahedron Lett., 2007, 48, 3299 CrossRef CAS;
(d) J. M. Khurana and S. Kumar, Tetrahedron Lett., 2009, 50, 4125 CrossRef CAS;
(e) M. M. Heravi, S. Sadjadi, N. M. Haj, H. A. Oskooie and F. F. Bamoharram, Catal. Commun., 2009, 10, 1643 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08111h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.