
Model system study of environmentally persistent free radicals formation in a semiconducting polymer modified copper clay system at ambient temperature†
Abstract
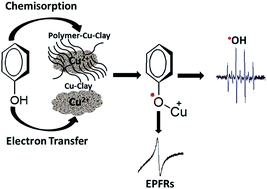
This paper systematically investigates how environmentally persistent free radicals (EPFRs) are formed in a phenol contaminated model soil. Poly-p-phenylene (PPP) modified and copper-loaded montmorillonite (MMT) clays were developed and used as models of soil organic matter and the clay mineral component, respectively, with phenol being employed as a precursor pollutant. The polymer modification of the clays was carried out via surface-confined Kumada catalyst-transfer chain-growth polymerization. The presence and location of the polymer were confirmed by a combination of thermogravimetric analysis (TGA), Raman spectroscopy, and X-ray diffraction data. EPFRs were formed by the Cu(II)-clay (Cu(II)CaMMT) and poly-p-phenylene–Cu(II)clay (PPP–Cu(II)CaMMT) composite systems under environmentally relevant conditions. The g-factor and concentration of EPFRs formed by the Cu(II)CaMMT and PPP–Cu(II)CaMMT systems were found to be 2.0034 and 1.22 × 1017 spins per g and 2.0033 and 1.58 × 1017 spins per g, respectively. These g-factors are consistent with the formation of phenoxyl radicals. Extended X-ray absorption fine structure (EXAFS) analysis shows that there are distinct differences in the local structures of the phenoxyl radicals associated with only the Cu(II) redox centers and those formed in the presence of the PPP polymer. X-ray absorption near edge spectroscopy (XANES) results provided evidence for the reduction of Cu(II) to Cu(I) in the EPFR forming process. The 1/e lifetimes of the formed EPFRs revealed a decay time of ∼20 h for the Cu(II)CaMMT system and a two-step decay pattern for the PPP–Cu(II)CaMMT system with decay times of ∼13.5 h and ∼55.6 h. Finally, the generation of reactive oxygen species (hydroxyl radical; ˙OH) by these clay systems was also investigated, with higher concentrations of ˙OH detected for the phenol-dosed Cu(II)CaMMT and PPP–Cu(II)CaMMT systems, compared to the non-EPFR containing undosed PPP–Cu(II)CaMMT system.
 Please wait while we load your content...
Please wait while we load your content...

