
Effect of ions on sorption of tylosin on clay minerals
Qian Zhangab,
Xiaohua Shuc,
Xuetao Guod,
Deqing Moa,
Shiguang Weia and
Chen Yang*b
aSchool of Life and Environmental Science, Guilin University of Electronic Technology, Guilin, Guangxi 541000, China
bCollege of Environmental Science and Engineering, South China University of Technology, Guangzhou, Guangdong 510006, China. E-mail: cyanggz542@gmail.com; Tel: +86 13632298598
cCollege of Environmental Science and Engineering, Guilin University of Technology, Guilin, Guangxi 541000, China
dSchool of Earth and Environment, Anhui University of Science and Technology, Huainan, Anhui 232001, China
First published on 16th May 2016
Abstract
A widely used veterinary antibiotic tylosin is associated with a fast increase in the prevalence of genes for macrolide resistance. Therefore, there are growing concerns of its potentially adverse effects on natural ecosystems, and a study on the sorption of tylosin is an important step in this direction. As a weak base, prior studies pointed out that cation exchange was the main sorption mechanism for tylosin in soil, and minerals were important factors in influencing the sorption. However, the ionic effect on the tylosin sorption process on minerals has not been systematically conducted. In this study, the sorption of tylosin on pure clay minerals was investigated in different ionic strength solutions. Moreover, the interlayer interaction of tylosin on different types of montmorillonite was preliminarily studied. All of the sorption data were fitted with the Langmuir–Freundlich–Hill sorption model. The results showed that the variation tendency of tylosin sorption capacity on montmorillonite and kaolinite were nearly the same. The sorption capacity of tylosin at different ionic strengths decreased with the order: 0.008 M > 0.051 M > 0.108 M > 0.508 M. In different ion solutions, the sorption capacity of tylosin decreased in the following order: K+ > Na+ > Ca2+ ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) Mg2+. However, the sorption of tylosin on montmorillonite at low initial concentration of tylosin displayed a different tendency. When the initial concentration of tylosin was low, its sorption capacities on montmorillonite at different ionic strengths decreased with the order: 0.051 M 0.108 M > 0.008 M > 0.508 M. For different ionic solutions, the sorption of tylosin decreased in the order of Mg2+ Ca2+ > K+ > Na+. The cation effect on tylosin sorption in the interlayers of montmorillonite was also investigated. The sorption of tylosin on different montmorillonites decreased in the order of Ca-montmorillonite > Na-montmorillonite > K-montmorillonite. These indicated that the hydration of inorganic ions influenced the sorption of tylosin. The results provide a more comprehensive understanding about the sorption of tylosin on minerals.
Mg2+. However, the sorption of tylosin on montmorillonite at low initial concentration of tylosin displayed a different tendency. When the initial concentration of tylosin was low, its sorption capacities on montmorillonite at different ionic strengths decreased with the order: 0.051 M 0.108 M > 0.008 M > 0.508 M. For different ionic solutions, the sorption of tylosin decreased in the order of Mg2+ Ca2+ > K+ > Na+. The cation effect on tylosin sorption in the interlayers of montmorillonite was also investigated. The sorption of tylosin on different montmorillonites decreased in the order of Ca-montmorillonite > Na-montmorillonite > K-montmorillonite. These indicated that the hydration of inorganic ions influenced the sorption of tylosin. The results provide a more comprehensive understanding about the sorption of tylosin on minerals.
1. Introduction
The presence of antibiotics in the environment is of growing interest worldwide due to the emergence and development of antibiotic resistance,1–3 which is considered as a potential risk for humans and the environment.4,5 Antibiotics are used extensively in veterinary medicine for therapeutic purposes and as a growth promoter. Veterinary antibiotics can be metabolized within the treated animal and eliminated from the body of the animal via urine and feces. However, 50–80% of antibiotics are excreted as unmetabolized compounds,6 becoming associated with manure and wastewater. So essentially all antibiotics administered are eventually excreted, whether unchanged or in metabolite form. The application of animal manure to agricultural fields as fertilizer is an important cause of the contamination of terrestrial environments by antibiotic residues and their biologically active metabolites. Although, the degradation of veterinary antibiotics can occur during manure storage, a large amount of then can still reach the soil environment via manure application.7 Additionally, these substances and their metabolites can enter aquatic environments, depending on their mobility in the soil system. This causes serious threats to both surface and groundwater quality and living organisms as a result of leaching from agricultural fields.8–18 They have been widely detected in surface water, groundwater, and soil.8,9,11Tylosin is a macrolide group antibiotic. It is widely used in poultry and swine feed as a growth promoter. Its use is related with the fast increase in the prevalence of genes for macrolide resistance, including resistance to an important macrolide human antibiotic erythromycin.19 In contrast with the direct interaction of people and livestock, the spreading of animal waste on agricultural fields was considered as an important pathway from this growth promoter leading to the occurrence of macrolide resistance in human pathogens.
Tylosin with a pKb value of 7.120 exists predominantly as a cationic species TYL+ at typical pH ranges of the natural environment. The log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kow value and water solubility of tylosin are 1.63 and 5 g L−1, respectively. The low Kow value and high water solubility suggest that tylosin is hydrophilic. Prior studies showed that antibiotics exhibited sorption properties that were very different from hydrophobic organic pollutants.21–23 As a weak base, the protonated tylosin could be bonded on negatively charged sites of soils and it is largely retained in soils and sediments.24 On the basis of the properties of tylosin, previous work pointed out that cation exchange was the main sorption mechanism for tylosin in soil.25 Tylosin demonstrates a high affinity for minerals. Bewick had reported that the distribution coefficients KD values of tylosin were about 8 to 128 L kg−1 on sandy loams and clay soils and that there was a positive relationship with clay content.26 Essington reported the sorption of tylosin on clays and clearly showed the effect of pH and ionic strength on the sorption of tylosin.27 However, detailed sorption mechanisms of tylosin have not been systematically conducted. Cation exchange and surface sorption will contribute significantly to the sorption of tylosin and provide a good explanation for the stronger sorption of tylosin on montmorillonite than that on kaolinite.28 Moreover, van der Waals forces also could play an important role on the distribution of tylosin. Loke et al. (2002) had reported that the distribution of tylosin in manure was predominantly related to hydrophobic binding to organic matter instead of ionic bonding formed with the aliphatic amine of the molecule.29
Kow value and water solubility of tylosin are 1.63 and 5 g L−1, respectively. The low Kow value and high water solubility suggest that tylosin is hydrophilic. Prior studies showed that antibiotics exhibited sorption properties that were very different from hydrophobic organic pollutants.21–23 As a weak base, the protonated tylosin could be bonded on negatively charged sites of soils and it is largely retained in soils and sediments.24 On the basis of the properties of tylosin, previous work pointed out that cation exchange was the main sorption mechanism for tylosin in soil.25 Tylosin demonstrates a high affinity for minerals. Bewick had reported that the distribution coefficients KD values of tylosin were about 8 to 128 L kg−1 on sandy loams and clay soils and that there was a positive relationship with clay content.26 Essington reported the sorption of tylosin on clays and clearly showed the effect of pH and ionic strength on the sorption of tylosin.27 However, detailed sorption mechanisms of tylosin have not been systematically conducted. Cation exchange and surface sorption will contribute significantly to the sorption of tylosin and provide a good explanation for the stronger sorption of tylosin on montmorillonite than that on kaolinite.28 Moreover, van der Waals forces also could play an important role on the distribution of tylosin. Loke et al. (2002) had reported that the distribution of tylosin in manure was predominantly related to hydrophobic binding to organic matter instead of ionic bonding formed with the aliphatic amine of the molecule.29
Although clays are often important sorbents for polar and ionizable organic compounds, limited studies of tylosin sorption have focused on pure clays. Indeed, clay minerals are widespread in soil and are a major factor to impact the fate and transport of organic contaminants. The pH and ionic strength play an important role on the sorption of tylosin on minerals.25,27 In the environment, the effect of ions is significant, especially for river estuaries, where the change of salinity may influence the environmental behavior of pollution. Thereby, following our study about the pH effect on tylosin adsorption on montmorillonite and kaolinite,30 this study focuses on the effect of solution ions. The solution ionic strength and ionic type were investigated in the tylosin sorption process on montmorillonite and kaolinite. Also, the interlayer interaction of tylosin on different types of montmorillonite were investigated. These experiments could help us to more fully understand the effect of ions on tylosin sorption, and sorption mechanism, including the interlayer interaction mechanism.
2. Materials and methods
2.1 Chemicals
All chemicals were analytical-reagent grade or higher purity and solvents were HPLC grade. Tylosin tartrate (TYL, >95%) was purchased from Sigma-Aldrich (St. Louis, MO) and used as received. Other chemicals including KNO3, NaNO3, Ca(NO3)2, Mg(NO)3, NaH2PO4, Na2HPO4, KH2PO4, NaOH, CaCl2, KCl and HNO3 were purchased from Guangzhou Chemical Reagent Co. (Guangzhou, China). Acetonitrile and formic acid were purchased from Merck (Darmstadt, Germany). Water was purified using Milli-Q® (Millipore, Guangzhou, China). The quality of the purified water was as follows: electrical resistivity = 18.2 MΩ cm TC; temperature = 24.9 °C, TOC = 4 μg L−1.A primary stock solution of tylosin at 1000 mg L−1 was prepared with Milli-Q® water and stored at 4 °C. The initial aqueous solutions were prepared by diluting desired volumes of the stock solution with pure water.
2.2 Sorbents
Montmorillonite and kaolinite purchased from The Clay Minerals Society (Chantilly, VA) were employed as the sorbents. Potassium- or calcium-saturated forms of the montmorillonite was prepared by wet sedimentation and subsequently exchanged with K+ or Ca2+. 10 g montmorillonite with 100 ml of either 0.1 M potassium chloride or calcium chloride was shaken for 4 h at 125 rpm and 25 ± 2 °C. Then the supernatant was removed after standing. This procedure was repeated four times until the cation exchange sites were saturated with K+ or Ca2+. Following this, the montmorillonite was washed with distilled water at least three times to remove excess salts. Finally the sample was subsequently quick-frozen and freeze-dried.2.3 Sorption and experiments
A batch reactor system using 40 ml glass centrifuge tubes with Teflon lined septa as the reactors was employed to measure equilibrium sorption of TYL on the sorbents. Preliminary tests were conducted to determine the optimal sorbent-to-solution ratio for achieving 30–70% reduction of the initial aqueous phase TYL concentrations (Co). In these tests, the Co levels were at 0.5, 10, 50 mg L−1 and the equilibration time was 24 h. A second preliminary test was performed to show that a mixing time of 72 h was sufficient to reach an apparent equilibrium.In the final tests, predetermined amounts of sorbents were weighed and placed into the reactors. After adding the initial aqueous TYL solution, the reactors were sealed immediately with the caps and wrapped with aluminum foil to minimize potential photodegradation. The reactors were then placed in a rotary shaker (125 rpm) at room temperature (25 ± 2 °C).
After equilibration, the reactors were centrifuged at 1400 g for 10 min, and an aliquot of the supernatant (approximately 1 ml) was transferred from each reactor to a pre-cleaned 1.5 ml amber glass vial for chemical analysis.
Ionic strength effect on tylosin sorption to clays was assessed by sorption isotherms. In this case, the solution pH was maintained constant at pH 6.5, while the ionic strength of background solution (KNO3) was varied from 0.008 to 0.508 M. The sorption isotherm was measured at a fixed pH 6.5 and different ionic strength background solution. Duplicate tubes were prepared at each of nine tylosin concentrations from 0.5 to 50 mg L−1.
In the case of the sorption isotherms of different ions, the solution pH and the ionic strength of solution were kept at pH 6.5 and 0.011 M, respectively. The background electrolytes were different and prepared from KNO3, NaNO3, Ca(NO3)2 and Mg(NO3)2. On the basis of the background electrolytes, the sorption isotherms were divided into four groups. In each group, duplicate tubes were prepared at each of the nine tylosin concentrations from 0.5 to 50 mg L−1.
The sorption isotherms of tylosin on K-montmorillonite, Ca-montmorillonite and Na-montmorillonite were measured at initial tylosin concentrations from 0.5 to 50 mg L−1. All of the sorption solutions were prepared using pure water. The solution pH of each tube was measured before and after sorption equilibrium.
2.4 Chemical analysis
The concentrations of tylosin in both the initial aqueous solutions and the supernatants were measured using a reverse-phase high performance liquid chromatograph (HPLC) (Agilent 1200) with C18 column (5 μm, 4.6 × 250 mm). This instrument was equipped with a diode array UV detector (wavelength at 290 nm for TYL). The mobile phase (flow rate 0.5 ml min−1) used was a mixture of acetonitrile and KH2PO4 solution (0.01 mol L−1) at a volumetric ratio of 35:65. External standards of tylosin (0.1–100 mg L−1) were used to measure a linear calibration curve. The solid-phase sorbate concentrations were computed based on the mass balance of the solute between the two phases.
2.5 Modeling
The equilibrium sorption data obtained for three clays and tylosin were fitted to the Langmuir–Freundlich–Hill (LFH) sorption model having the following form:where qe is the equilibrium solid-phase solute concentration (mg kg−1), Ce is the aqueous-phase solute concentration (mg L−1), Km is the LFH sorption coefficient, n is the isotherm nonlinearity index (unitless). Parameter n is a measure of the heterogeneity of the binding surface.
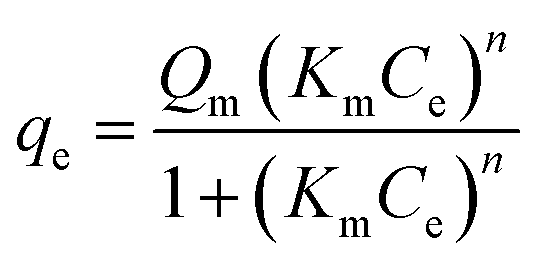
When the affinity does not depend on the concentration or the binding, then n = 1; when the binding shows a positive cooperativity (hydrophobic interaction), the affinity increases with increasing coverage and/or concentration n > 1. In this case, the equation is called the Hill equation. When the binding occurs on sites that are heterogeneous, the affinity gradually decreases because sites with high affinity will be preferentially occupied at low concentration and the low affinity sites will be occupied at high concentration, and n < 1. In this case, we have the so-called Langmuir–Freundlich equation.
The LFH model parameters were obtained with a nonlinear regression approach using the computer software (Origin 8.5, OriginLab Co.).
3. Results and discussion
3.1 Effect of ionic strength on sorption
The tylosin sorption isotherms at different ionic strength solutions for montmorillonite and kaolinite are shown in Fig. 1. The best fit model of LFH and the fitting parameters are summarized in Table 1. The results obviously showed that the maximum sorption capacities Qm of tylosin on the two minerals were decreased with increasing ionic strengths. The fitting parameter n value was in the range of 0.901–1.332 and 0.620–1.237 for montmorillonite and kaolinite, respectively. Most of the n values was less than 1 except for the sorption at 0.1 M ionic strength. | ||
| Fig. 1 TYL sorption isotherms at different ion strengths. | ||
| Sample | Ion strength | Sorption | |||
|---|---|---|---|---|---|
| Qm/mg kg−1 | Km | n | R2 | ||
| MON | 0.008 M | 426832.997 |
0.042 | 0.902 | 0.988 |
| 0.051 M | 184721.428 |
0.441 | 0.997 | 0.976 | |
| 0.108 M | 123864.254 |
1.332 | 1.188 | 0.962 | |
| 0.505 M | 67305.455 |
1.269 | 0.901 | 0.963 | |
| KAO | 0.008 M | 749.682 | 0.126 | 0.620 | 0.988 |
| 0.051 M | 525.558 | 0.052 | 0.628 | 0.986 | |
| 0.108 M | 141.381 | 0.720 | 1.237 | 0.979 | |
| 0.505 M | 113.098 | 0.627 | 0.864 | 0.990 | |
Fig. 1 shows that the sorption of tylosin on kaolinite was decreased with increased ionic strength at all the solution concentrations. However, the ionic strength effect on the sorption of tylosin on montmorillonite was different at low and high concentrations of tylosin. When the initial concentration was high, the sorption capacity of tylosin at different ionic strengths decreased with the order: 0.008 M > 0.051 M > 0.108 M > 0.508 M. The sorption decreased obviously with increasing ionic strength. However, when the initial solution was low, the sorption capacities at ionic strengths of 0.051 and 0.108 M were nearly the same. However they were larger than the sorption capacity at the ionic strength of 0.008 M.
The different sorption performance of tylosin on montmorillonite could be caused by the heterogeneous sorption sites of montmorillonite. When the initial concentration of tylosin was low, the molecules first occupied high affinity sites via hydrogen bonding. Tylosin can interact with the mineral surface groups as well as the water molecules. In general, K+ in solution can compete with TYL+ for negatively charged sites on the clay surface and this may cause the decrease of sorption with increased ionic strength. However, the increased ionic strength promoted the sorption over a reasonable range of ionic strength. This can be explained by there being a lot of water around the K+ attracted by the negative sites. The tylosin may interact with OH of water to increase the sorption at a reasonable range of ionic strength. Once the ionic strength was out of this range, the competition of K+ for the negatively charge sites caused an obvious decrease of tylosin sorption. Therefore the sorption of tylosin at high ionic strength was weakest.
When the initial concentration of tylosin was high, tylosin interacted with the low energy sorption sites on montmorillonite and cation exchange was the major interaction. Sorption of tylosin by cation exchange should be accompanied by the release of inorganic cations associated with cation exchange sites of the clay surface, e.g., Na+ for Na-MON. As such, the addition of cationic species released by cation exchange at increasingly higher concentrations with cationic tylosin should cause a shift in the cation exchange equilibrium toward reduced sorption of tylosin. The maximum aqueous equilibrium concentration of tylosin found experimentally was 0.049 mmol L−1. Even at a concentration of K+ of 0.01 M, this is 200 times greater than the concentration of tylosin in aqueous solution. Such large discrepancies between K+ and tylosin concentration in solution phase could cause cation exchange equilibration toward less release of Na+ from the clay surface, and this significantly reduced sorption for tylosin is in accordance with Le Chatelier’s principle. The presence of relatively high ionic strength of inorganic cations could effectively compete with trace levels of tylosin for sorptive sites on mineral surfaces, so reducing the sorption of tylosin.
The sorption of tylosin on kaolinite decreased with increased ionic strength at all concentrations. The positively charged ions are attracted to negatively charged sites on the surface of kaolinite by electrostatic attraction. The competition between TYL+ and cations in the solution resulted in a decreased sorption of TYL on kaolinite. Fig. 2 plots the zeta potential against the solution ionic strength and illustrates the charge change on mineral surface. From Fig. 2, it was apparent that the negative charge on the surface decreased gradually with the increased ionic strength. This directly proved that inorganic cations in solutions attach to some of the negative sites on the mineral surface. However, there was a sharp decrease in the zeta potential of montmorillonite at low ion strength. This may be related with the reverse flocculation of montmorillonite. Clay particles can aggregate through edge to edge, face to face and face to edge interactions according to the Gouy–Chapman double layer model. However, reverse aggregation may occur when the concentration of electrolytic is low. The compression of the double layer may reduce the attraction between the edge and face and may favor the dispersion of montmorillonite.
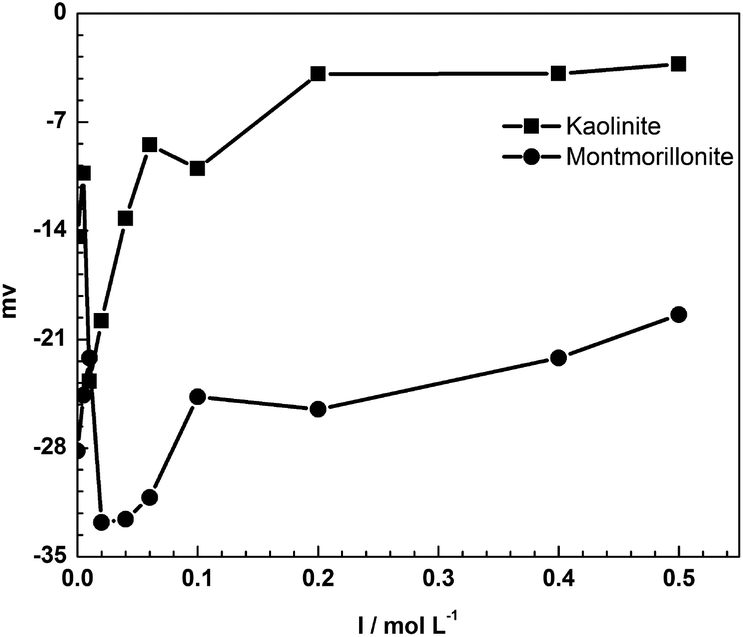 | ||
| Fig. 2 Zeta potential of minerals at different ionic strengths. | ||
The FLH fitting parameter n reflects the change of interaction force between the sorption site and sorbate; n < 1 indicated the force between tylosin and the sorption site decreased with increasing concentration and indicated tylosin first occupied the high energy sites at low concentration. As the concentration increased, tylosin interacted with lower energy sites gradually. So the sorption sites of the clay was heterogeneous and the sorption isotherms were nonlinear. The results were consistent with the information provided from Fig. 1. When n > 1 a hydrophobic interaction is also suggested.
3.2 Effect of ion solutions on the sorption
To further evaluate the solution ion effects on tylosin sorption on minerals, the effect of ionic type in solution was investigated. The sorption isotherms for different ion solutions are plotted in Fig. 3. This showed that the sorption of tylosin on kaolinite decreased in the order of K+ > Na+ > Ca2+ Mg2+ at all initial concentrations of tylosin. However, the sorption of tylosin on montmorillonite was more complicated than that on kaolinite. Specifically, sorption of tylosin for different ionic solutions at pH 6.5 decreased in the order of Mg2+ Ca2+ > K+ > Na+ at low initial concentration of tylosin, while at high initial concentration of tylosin, the sorption in univalent inorganic cation solutions was stronger than that at divalent inorganic cation solution; the decreasing order was: K+ > Na+ > Ca2+ Mg2+.
 | ||
| Fig. 3 TYL sorption isotherms for different ionic solutions. | ||
Sorption coefficients were calculated from the sorption isotherms fitted with the FLH model and the results are given in Table 2. Tylosin equilibrium sorption capacities Qm in different ionic solutions followed the order of K+ > Na+ > Ca2+ > Mg2+ for kaolinite and K+ > Na+ > Mg2+ > Ca2+ for montmorillonite. The n value for kaolinite was <1 and the n value for montmorillonite was >1 except for Mg2+.
| Sample | Ion | Sorption | |||
|---|---|---|---|---|---|
| Qm/mg kg−1 | Km | n | R2 | ||
| MON | K+ | 305081.474 |
0.175 | 1.452 | 0.990 |
| Na+ | 247478.929 |
0.145 | 2.442 | 0.980 | |
| Ca2+ | 187301.042 |
1.600 | 1.236 | 0.975 | |
| Mg2+ | 206327.402 |
0.921 | 0.868 | 0.986 | |
| KAO | K+ | 658.904 | 0.096 | 0.695 | 0.989 |
| Na+ | 324.900 | 0.436 | 0.766 | 0.987 | |
| Ca2+ | 311.495 | 0.154 | 0.690 | 0.975 | |
| Mg2+ | 251.041 | 0.345 | 0.804 | 0.990 | |
The assumed model was that the mineral surface was heterogeneous with different energy sorption sites. It can be used to explain the different sorption performance of tylosin on montmorillonite for the four inorganic ion solutions. At low initial concentrations of tylosin, tylosin first took up high energy sites to complete surface sorption. The strong hydration of the inorganic cation attracts more water molecules surrounding the cation. Tylosin molecules could interact with –OH of water, and divalent inorganic cations (Mg2+, Ca2+) with stronger hydration energy may promote the sorption of tylosin. In fact, Xu and Boyd have reported the cation surfactant hexadecyltrimethylammonium (HDTMA) sorption by montmorillonite manifested a similar sorption order: Ca-MON > Cs-MON > Na-MON.31 The authors reported that HDTMA sorption via cation exchange interaction with clays also involved favorable lateral interactions among the hydrophobic C-16 alkyl moieties of exchanged HDTMA. Na-smectite with an initially dispersed state reduced such lateral interaction and hence disfavored HDTM sorption compared to more flocculated clays such as Cs- and Ca-smectite. The similar sorption phenomena may indicate interaction between tylosin molecules are similar to HDTMA sorption on montmorillonite. Previous study about the pH effect on tylosin sorption on minerals have pointed out adsorption of tylosin on minerals could increase the organic content of mineral. And as a part of the mineral, they may interact with tylosin in the solution to promote the sorption of tylosin.30 An LFH fitting parameter n > 1 also indicates the possible hydrophobic interaction between tylosin molecules. At a high initial concentration of tylosin it will bind to low energy sorption sites. Cation exchange is the likely major interaction under such conditions. Divalent cations (Ca2+, Mg2+) bind more favorably by electrostatic attraction relative to univalent cations (K+, Na+) and it is difficult for TYL+ to compete for the negative charge sites on montmorillonite with divalent cations (Ca2+, Mg2+). So the sorption at univalent inorganic cation solution was stronger than that at divalent inorganic cation solution at high initial concentration of tylosin.
Electrostatic attraction causes the competition of negative charge sites between TYL+ and inorganic cations on kaolinite. Hence, the sorption decreased in the order of K+ > Na+ > Ca2+ ≅ Mg2+. The stronger sorption in K+ solution, relative to Na+ solution in the cation exchange process, may be related to the lower hydration energy of K+.
3.3 Sorption of tylosin on different ionic montmorillonites
Tylosin can enter the interlayer of montmorillonite but the question is whether this is through cation exchange and whether the cation in the interlayer affects the sorption of tylosin? To answer these questions, different types of montmorillonite were prepared by saturating with exchangeable cations (K+, Ca2+) and the sorption isotherms were examined. Sorption isotherms in Fig. 4 showed tylosin sorption on montmorillonite decreased in the order of Ca-montmorillonite > Na-montmorillonite > K-montmorillonite. LFH fitting results Qm in Table 3 were in the same order. | ||
| Fig. 4 TYL sorption isotherms for different ionic montmorillonites. | ||
| Sample | Sorption | |||
|---|---|---|---|---|
| Qm/mg kg−1 | Km | n | R2 | |
| K-MON | 67727.695 |
0.509 | 2.314 | 0.960 |
| Na-MON | 170882.924 |
0.593 | 0.82 | 0.970 |
| Ca-MON | 2490089.233 |
7.761 × 10−6 | 0.279 | 0.854 |
The sorption solution was pure water in this experiment. Hence no cation competed with TYL+ on the mineral surface. If the major factor for the tylosin sorption process was the cation binding strength of the interlayer of montmorillonite, then tylosin exchange with the divalent Ca2+ cation, which strongly interacts with negatively charged sites of the mineral through electrostatic interaction, should be more difficult compared to univalent cations Na+ or K+. So the sorption of tylosin on Ca-montmorillonite should be the weakest. However, this was contrary to the experiment result. We should also consider the surface charge equilibrium of montmorillonite through the zeta electric potential. The zeta electric potential of Ca-montmorillonite and Na-montmorillonite were −17 and −33 mV before adsorption of tylosin. After adsorption, the zeta electric potential of Ca-montmorillonite was −20 mV, and the zeta electric potential of Na-montmorillonite was −28 mV. The decreased zeta electric potential of Ca-montmorillonite may increase the sorption of tylosin. However, the zeta electric potential of Ca-montmorillonite was higher than the Na-montmorillonite whether before or after adsorption. If the zeta electric potential was considered only, the sorption quantity of Na-montmorillonite should be higher than Ca-montmorillonite. Therefore, the interlayer hydration may be important for tylosin sorption on Ca-montmorillonite rather than the cation exchange and surface charge. The hydration energy for K+ is −321 kJ mol−1, less than that of Na+ (−405 kJ mol−1) and much less than that of Ca2+ (−1592 kJ mol−1). The relatively strong hydration of Ca2+ attracts more water, leading to larger expansion of montmorillonite layer. This was proved by the XRD results of the three montmorillonites in Fig. 5 which shows the basal spacing of Ca-montmorillonite was 1.38 nm, larger than for K-montmorillonite and Na-montmorillonite (1.15 and 1.13 nm). Ca2+ attracted more water in the interlayer and promotes tylosin to interact with OH of water via hydrogen bonding. Hence more tylosin molecules were adsorbed on Ca-montmorillonite. High valent cations in the interlayer of montmorillonite can therefore lead to stronger sorption ability for some organic materials. Similar reports have appeared previously; Sawhney and Singh reported that the sorption of atrazine on Al-montmorillonite was far higher than that on Ca-montmorillonite.32 Pusino et al. studied the dimepiperate sorption on Na+, Ca2+, Fe3+, Al3+ montmorillonites and the sorption capacity increased in the order of Na+ < Ca2+ < Al3+ < Fe3+.33 The NMR measurement of pyridine sorption on montmorillonite proved that hydrogen bonds were generated in the sorption process.34 Aside from the hydrogen bonding discussed above, the interaction between tylosin molecules in the interlayer may increase the adsorption of tylosin and develop “hydrophobic barriers”. This may hamper the release of exchangeable inorganic cations from the interlayer.
 | ||
| Fig. 5 XRD analyses for different montmorillonites. | ||
While the basal spacing for Na-montmorillonite and K-montmorillonite is similar in Fig. 5, the actual basal spacing of Na-montmorillonite in solution is much larger than K-montmorillonite due to the higher hydration energy of Na+ (−405 kJ mol−1) relative to K+ (−321 kJ mol−1). The adsorption of tylosin on the three montmorillonites increased in the order of Ca-montmorillonite > Na-montmorillonite > K-montmorillonite. This result was consistent with the theoretical interlayer spacing and suggested that cation exchange may be negligible in the montmorillonite interlayer. Consideration of hydration energies and hydrogen bonds formed between tylosin and interlayer water may be the best explanation for the adsorption results of tylosin on the three montmorillonites.
The sorption of tylosin on different types of montmorillonite indicated that interlayer hydration was an important factor to influence the sorption of tylosin. Not only hydration affected the interlayer cation exchange, but also the hydrogen bonds formed between tylosin and interlayer water was more important than cation exchange in the montmorillonite interlayer.
4. Conclusions
Low ionic strength and low valent cations in solution promoted the sorption of tylosin on clays and proved that cation exchange was important in the sorption process. However, the sorption mechanism of tylosin on montmorillonite was more complicated. It was influenced by the concentration of tylosin, the hydration state of clay surface and interlayer. Tylosin adsorption on montmorillonite displayed an opposite tendency at low initial concentration of tylosin and indicated that the sorption sites on clays were heterogeneous. When tylosin and ions coexist in solution, tylosin first occupied high energy sites via hydrogen bonding at a relative low concentration and hydrophobic binding between tylosin molecules may also exist. As the concentration increased, they gradually occupied the lower energy sites via cation exchange. Not only the cations in solution can compete with positive charged tylosin ions, but also the hydration of cations obviously influenced the sorption. In the interlayer, the hydration status of the montmorillonite interlayer appears to be more important than the interlayer cation exchange.Acknowledgements
The study was financially supported by the China National Science Fund Program (No. 41501342; No. 41503095; No. 31460155) and Guangxi Natural Science Foundation (2014GXNSFAA118324).References
- P. A. Blackwell, A. B. A. Boxall, P. Kay and H. Noble, J. Agric. Food Chem., 2005, 53, 2192–2201 CrossRef CAS PubMed.
- P. A. Blackwell, P. Kay and A. B. A. Boxall, Chemosphere, 2007, 67, 292–299 CrossRef CAS PubMed.
- P. A. Blackwell, P. Kay, R. Ashauer and A. B. A. Boxall, Chemosphere, 2009, 75, 13–19 CrossRef CAS PubMed.
- A. R. Khachatryan, T. E. Besser, D. D. Hancock and D. R. Call, Appl. Environ. Microbiol., 2006, 72, 4583–4588 CrossRef CAS PubMed.
- A. K. Sarmah, M. T. Meyer and A. B. A. Boxall, Chemosphere, 2006, 65, 725–759 CrossRef CAS PubMed.
- E. Vaclavik, B. Halling-Sørensen and F. Ingerslev, Chemosphere, 2004, 56, 667–676 CrossRef CAS PubMed.
- F. Ingerslev and B. Halling-Sørensen, Ecotoxicol. Environ. Saf., 2001, 48, 311–320 CrossRef CAS PubMed.
- G. Hamscher, H. T. Pawelzick, H. Höper and H. Nau, Environ. Toxicol. Chem., 2005, 24, 861–868 CrossRef CAS PubMed.
- G. Hamscher, S. Sczesny, H. Höper and H. Nau, Anal. Chem., 2002, 74, 1509–1518 CrossRef CAS PubMed.
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman, S. D. Zaugg, L. B. Barber and H. T. Buxton, Environ. Sci. Technol., 2002, 36, 1202–1211 CrossRef CAS PubMed.
- E. Zuccato, S. Castiglioni and R. Fanelli, J. Hazard. Mater., 2005, 122, 205–209 CrossRef CAS PubMed.
- J. G. Davis, C. C. Truman, S. C. Kim, J. C. Ascough and K. Carlson, J. Environ. Qual., 2006, 35, 2250–2260 CrossRef CAS PubMed.
- K. Stoob, H. P. Singer, S. R. Mueller, R. P. Schwarzenbach and C. H. Stamm, Environ. Sci. Technol., 2007, 41, 7349–7355 CrossRef CAS PubMed.
- X. Peng, J. Tan, C. Tang, Y. Yu and Z. Wang, Environ. Toxicol. Chem., 2008, 27, 73–79 CrossRef CAS PubMed.
- A. Hoese, S. A. Clay, D. E. Clay, J. Oswald, T. Trooien, R. Thaler and C. G. Carlson, J. Environ. Sci. Health, Part B, 2009, 44, 371–378 CrossRef CAS PubMed.
- Y. Luo, L. Xu, M. Rysz, Y. Wang, H. Zhang and P. J. J. Alvarez, Environ. Sci. Technol., 2011, 45, 1827–1833 CrossRef CAS PubMed.
- W. Xu, G. Zhang, X. Li, S. Zou, P. Li, Z. Hu and J. Li, Water Res., 2007, 41, 4526–4534 CrossRef CAS PubMed.
- Y. W. Li, X. L. Wu, C. H. Mo, Y. P. Tai, X. P. Huang and L. Xiang, J. Agric. Food Chem., 2011, 59, 7268–7276 CrossRef CAS PubMed.
- F. M. Aarestrup and B. Carstensen, Microb. Drug Resist., 1998, 4, 307–312 CrossRef CAS PubMed.
- L. Wollenberger, B. Halling-Sørensen and K. O. Kusk, Chemosphere, 2000, 40, 723–730 CrossRef CAS PubMed.
- A. B. A. Boxall, P. Blackwell, R. Cavallo, P. Kay and J. Tolls, Toxicol. Lett., 2002, 131, 19–28 CrossRef CAS PubMed.
- C. Gu and K. G. Karthikeyan, J. Environ. Qual., 2008, 37, 704–711 CrossRef CAS PubMed.
- S. Thiele-Bruhn, T. Seibicke, H. R. Schulten and P. Leinweber, J. Environ. Qual., 2004, 33, 1331–1342 CrossRef CAS PubMed.
- S. C. Kim and K. Carlson, Environ. Sci. Technol., 2007, 41, 50–57 CrossRef CAS PubMed.
- T. L. ter Laak, W. A. Gebbink and J. Tolls, Environ. Toxicol. Chem., 2006, 25, 904–911 CrossRef CAS PubMed.
- M. W. M. Bewick, Plant Soil, 1979, 51, 363–372 CrossRef CAS.
- M. E. Essington, J. Lee and Y. Seo, Soil Sci. Soc. Am. J., 2010, 74, 1577–1588 CrossRef CAS.
- Y. Seo, Ph.D, The University of Tennessee, 2006.
- M. L. Loke, J. Tjørnelund and B. Halling-Sørensen, Chemosphere, 2002, 48, 351–361 CrossRef CAS PubMed.
- Q. Zhang, C. Yang, W. Huang, Z. Dang and X. Shu, Chemosphere, 2013, 93, 2180–2186 CrossRef CAS PubMed.
- S. Xu and S. A. Boyd, Environ. Sci. Technol., 1995, 29, 3022–3028 CrossRef CAS PubMed.
- B. L. Sawhney and S. S. Singh, Clays Clay Miner., 1997, 45, 333–338 CAS.
- A. Pusino, W. Liu and C. Gessa, Clays Clay Miner., 1993, 41, 335–340 CAS.
- L. Ukrainczyk and K. A. Smith, Environ. Sci. Technol., 1996, 30, 3167–3176 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |