
Caffeic acid attenuates the angiogenic function of hepatocellular carcinoma cells via reduction in JNK-1-mediated HIF-1α stabilization in hypoxia†
Abstract
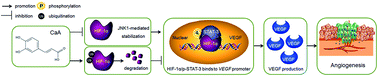
Hepatocellular carcinoma (HCC) is the third leading cause of tumor-related mortality worldwide. Angiogenesis plays a crucial role in HCC progression. Caffeic acid (CaA) is a novel anti-tumor agent, however, the functions of CaA in the regulation of angiogenesis in HCC, and the molecular mechanisms involved, remain largely uninvestigated. Here, we found that, in the presence of CoCl2 (a hypoxia mimic), CaA attenuates the angiogenic function of HCC cells via reduction in JNK-1-mediated HIF-1α stabilization. Briefly, CaA attenuated the CoCl2-induced autocrine vascular endothelial growth factor (VEGF) and angiogenesis in HCC cells. For the molecular mechanisms, CoCl2 treatment increased the expressions of HIF-1α and phosphorylated signal transducers and activators of transcription-3 (p-STAT-3). Then, by directly binding to the promoter of the VEGF gene, HIF-1α effectively activated VEGF. However, CaA attenuated the CoCl2-induced activation of HIF-1α likely by reducing JNK1 activation and reducing HIF-1α stabilization. Moreover, CaA decreased the CoCl2-induced increased expression of p-STAT-3. These two functions resulted in an attenuated recruiting of the HIF-1α to the VEGF promoter. By understanding a novel mechanism whereby CaA inhibits the angiogenesis in HCC, our study expands the understanding of the molecular mechanisms involved in the anti-cancer potential induced by CaA.
 Please wait while we load your content...
Please wait while we load your content...

