
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the mechanism and structural features of polysorbate 80 during adsorption on PLGA nanoparticles by attenuated total reflectance – Fourier transform infrared spectroscopy†
Abhayraj S.
Joshi
,
Avinash
Gahane
and
Ashwani Kumar
Thakur
*
Department of Biological Sciences and Bioengineering, Indian Institute of Technology, IIT Kanpur, Kanpur, Uttar Pradesh, India – 208016. E-mail: akthakur@iitk.ac.in
First published on 7th November 2016
Abstract
Polysorbate 80 coated nanoparticles have been studied extensively for delivery of many bioactive molecules to the brain for diagnosis and treatment of different neurological disorders. The adsorption mechanism of polysorbate 80 on nanoparticles is not known due to a lack of suitable techniques for quantification at low concentrations. Here we report the probable mechanism of polysorbate 80 adsorption on poly-D,L-lactide-co-glycolide (PLGA) nanoparticles using a novel extraction procedure coupled with a highly sensitive attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopic method. Both methods facilitated the characterization and quantification of extracted surfactant at the nanogram level. Adsorption isotherm modeling reveals that polysorbate 80 follows a physisorption process (ΔG = 38 kJ Mol−1) and forms a monolayer on the nanoparticle surface. Thermal analysis and X-ray photoelectron spectroscopy (XPS) further support polysorbate 80 coating and surface coverage on PLGA nanoparticles. Real-time ATR-FTIR analysis shows that polysorbate 80 interacts with nanoparticles through the ester group (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and acyl chain (–CH2) during the adsorption. The extent and direction of band shifts in real-time FTIR experiments suggest that the acyl chain of polysorbate 80 micelles loses its ordered structure and becomes flexible with a higher gauche/trans conformer ratio. Reduction in the degree of hydration of the ester group indicates an increase in hydrophobic interactions in the presence of PLGA nanoparticles. The results of FTIR analysis states that polysorbate 80 acquires a flat structure on the nanoparticle surface. This study on polysorbate 80 quantification and adsorption with ATR-FTIR spectroscopy can be extended to other non-ionic surfactants which will ultimately help in determination of biological fate of surfactant coated nanoparticles under in vivo conditions.
O) and acyl chain (–CH2) during the adsorption. The extent and direction of band shifts in real-time FTIR experiments suggest that the acyl chain of polysorbate 80 micelles loses its ordered structure and becomes flexible with a higher gauche/trans conformer ratio. Reduction in the degree of hydration of the ester group indicates an increase in hydrophobic interactions in the presence of PLGA nanoparticles. The results of FTIR analysis states that polysorbate 80 acquires a flat structure on the nanoparticle surface. This study on polysorbate 80 quantification and adsorption with ATR-FTIR spectroscopy can be extended to other non-ionic surfactants which will ultimately help in determination of biological fate of surfactant coated nanoparticles under in vivo conditions.
Introduction
Polysorbate 80, an oleic acid ester of polyethoxylated sorbitan, is a commonly used non-ionic surfactant. Fatty acid chains (hydrophobic) and ethylene oxide units (hydrophilic) offer an amphipathic nature to this molecule.1 Like other surfactants, polysorbate 80 also forms micelles in the concentration range of 0.0010–0.0015%.2–4 Owing to its amphipathic nature, polysorbate 80 has been used as a solubilizer of hydrophobic drugs,5 wetting agent,6 emulsifier,7 stabilizer of proteins8 in food, cosmetic and pharmaceutical industries.1,9 Apart from these roles, it is a potential coating agent for nanoparticulate formulations that are specifically designed to target the central nervous system (CNS).10 The literature shows that polysorbate 80 coating on nanoparticle surface improves CNS intake of encapsulated bioactive molecules and theranostic agents.11–13 Upon intravenous administration, polysorbate 80 coated nanoparticles adsorb apolipoprotein E (ApoE) and cross the blood–brain barrier (BBB) through low density lipoprotein receptors.14–17 The nanoparticles prepared from poly-D,L-lactide-co-glycolide (PLGA), a FDA approved biodegradable polymer, have shown great potential in drug delivery.18 Polysorbate 80 coated PLGA nanoparticles are in the research phase for delivering drugs to brain for different neuropathological conditions including Alzheimer's disease, Huntington's disease, Parkinson's disease and brain cancer.19–21 However, very few articles show the quantification of polysorbate 80 adsorbed on the surface of polymeric nanoparticles.14,22,23In general, adsorption of a surfactant on nanoparticles is a fundamental phenomenon and it is governed by different interactions with nanoparticle surface.24–26 The size and surface properties of nanoparticles may influence the amount, adsorption pattern and the adsorption kinetics of the surfactant. In case of polysorbate 80 coated PLGA nanoparticles, incomplete adsorption of the surfactant may lead to poor apolipoprotein adsorption and reduced target specificity.27 It has been shown that the hydrophilic as well as hydrophobic parts of non-ionic surfactants are actively involved in interactions with the cells.28 Coating with various non-ionic surfactants from poloxamer and polysorbate families directly suppress the action of reticuloendothelial system and thus help in prolonging the circulation time of nanoparticles in vivo.29 Also, the surfactant coating reduces the uptake by liver and spleen probably because of reduced non-specific protein adsorption on nanoparticles.17,29,30 Hence, the quantification and adsorption of polysorbate 80 is important to design the nanoparticulate drug delivery system with optimal surfactant coating needed for reducing the non-specific protein adsorption, improving brain targeting and prolonging the circulation time. Moreover, the adsorption mechanism for polysorbate 80 on PLGA nanoparticles has not been determined yet, because of lack of a sensitive method for quantification. For determining the concentration of polysorbate 80 in an aqueous solution, several methods are available. UV-visible spectrophotometry is the most widely accepted method for quantification. Fluorescence spectroscopy, liquid chromatography and gas chromatography are few other methods used for quantification of polysorbate 80. Recently, Fourier transform infra-red spectroscopic (FTIR) method has been developed and patented by Glaxosmithkline Biologicals S.A. (Belgium) for determination of polysorbate 80 in vaccines and protein solutions.31 UV-visible, fluorimetry and chromatographic methods have limitations with respect to sensitivity and applicability. The FTIR method can be useful for detection and quantification due to high specificity, robustness and capability of quantifying polysorbate 80 at nanogram level. Additionally, ATR-FTIR method has been previously used for adsorption studies; however, its applicability in adsorption studies on nanoparticles still remains unexplored.32 The nanoparticles may interfere with UV and fluorimetry based methods due to their light scattering properties which makes analysis of polysorbate 80 coated nanoparticles cumbersome. Therefore, it seems essential to extract polysorbate 80 from nanoparticle surface for the quantification and to understand its adsorption mechanism.
In this paper, we are reporting a novel and simple solvent extraction method for the removal of polysorbate 80 coating from the surface of PLGA nanoparticles. We have used differential scanning calorimetry (DSC) and X-ray photoelectron spectroscopy (XPS) to confirm the polysorbate 80 coat on the surface of PLGA nanoparticles. After extraction, we have quantified polysorbate 80 by ATR-FTIR method. We have extended the use of ATR-FTIR method for the determination of its adsorption kinetics and its interactions with surface of PLGA nanoparticles. By performing adsorption isotherm modeling and adsorption kinetic modeling, the adsorption mechanism is determined. By using real-time FTIR, we have deduced possible structural changes that polysorbate 80 undergoes during the adsorption process.
Results and discussion
Variety of nanocarrier systems containing various bioactive molecules have been engineered for the treatment of cancer, inflammatory conditions and genetic disorders.18 In particular, polysorbate 80 coated polymeric nanoparticles are in the preclinical phase for the diagnosis and the treatment of various neuropathological conditions including brain cancer18 and neurodegenerative diseases.33 The surface modification with polysorbate 80 is responsible for the interactions of nanoparticles with serum apolipoproteins, apolipoprotein adsorption, and receptor mediated endocytosis at BBB.11,14,33 Thus, understanding the mechanistic details underlying the process of surfactant adsorption can provide insights for predicting the behavior of nanoparticles in the presence of several intrinsic and extrinsic cellular players. In previous attempts, estimation of polysorbate 80 adsorbed on biodegradable nanoparticles was done using indirect UV-visible spectrophotometric method. However, adsorption mechanism was not identified. Also, no extraction procedure is available to remove the adsorbed amount of polysorbate 80 from PLGA nanoparticles. Therefore, we designed a protocol for extraction of polysorbate 80 from PLGA nanoparticles followed by its characterization using highly sensitive FTIR method. By using our previously optimized protocol of nanoprecipitation, PLGA nanoparticles of less than 200 nm were prepared. The results obtained from dynamic light scattering shows uniform size distribution and comply with our previously reported data.34Development of ATR-FTIR method for quantification of polysorbate 80
To quantify polysorbate 80 on nanoparticles, ATR-FTIR method was used. We first developed the standard curve of polysorbate 80 using different concentrations (Table S1†). All solutions were analyzed in liquid (a drop on ZnSe crystal) (Fig. 1) as well as dry state (a drop of sample air dried on ZnSe crystal) (Fig. S1†). All concentrations in both the physical states (solid and liquid) showed IR peaks (Table S2†) with three characteristic bands in the range of 3000–2800 cm−1, 1770–1720 cm−1 and 1200–1050 cm−1 representing –CH2 (acyl chain of fatty acid), –CO group of ester moiety and –C–O group from ethoxy moiety, respectively (Fig. S2 and S3†).
 | ||
| Fig. 1 (A) Standard curve for polysorbate 80 by ATR-FTIR method in liquid state. Stack of primary spectra for (B) acyl chain (–CH2) stretching vibrations, (C) ester group (–CO) stretching vibrations and (D) acyl chain scissoring vibrations. The black arrows in (C) indicate fusion of peaks. | ||
Among these peaks, the acyl chain peak was selected for the quantification of polysorbate 80 because of its intense band at very low concentration. Same band position has been used previously for adsorption and desorption studies of poly(ethylene glycol) monoalkyl ether based nonionic surfactant.35 Due to environmental influence, FTIR spectroscopic method is prone to significant spectral variations for the same sample that may create error during quantification. Hence, for normalization fixed concentration (1 mg ml−1) of sodium azide was used. Sodium azide is a suitable internal standard36 because it has no interference in the region where polysorbate 80 shows signature peaks (Fig. S4†).31 It gives a very sharp peak in the region of 2080–1970 cm−1 (in liquid state) and 2195–2095 cm−1 (in dry state). The change in peak position can be attributed to ionic interactions and hydrogen bonding of sodium azide in water.37 In solution form, azide ion forms hydrogen bonds with water molecules. In dry salt form, such interactions are absent.37 Using ratio of peak area of polysorbate 80 to peak area of sodium azide, normalization was done (calculation SC1†) and standard curves were plotted as area ratio (AreaAcyl chain/AreaAzide) versus different amount of polysorbate 80 put on the ATR crystal (Fig. 1 and S1†).
In the standard curve of polysorbate 80 solutions, linearity was found within the range of 1.25 ng to 20 ng. Below 1.25 ng, no detection was observed. On the other hand, for dried polysorbate 80 samples, the linearity range was from 0.3 to 20 ng. The limit of quantitation (LOQ) is 0.3 ng and the limit of detection (LOD) is in between 0.3 ng to 0.15 ng. In dry state, the thickness of polysorbate 80 on ATR crystal decreases and thus improves the internal reflections of IR beam. This ultimately leads to improvement of IR signal reaching to the detector. The inverse relationship between IR signal of a sample and its thickness has been previously reported.38 Thus, the LOD has lower value for dry polysorbate 80 as compared to polysorbate 80 solution. However, as a result of uneven and uncontrolled air drying, analysis of dry polysorbate 80 samples suffers with drawback of high variability in the results irrespective of better IR signal detection. On the other hand, the analysis of polysorbate 80 in liquid state showed higher reproducibility with low standard error. The percentage accuracy in analysis of liquid as well as dry polysorbate 80 samples lies within the range of 80–120% which is in compliance with the guidelines for analytical method development39 (Fig. S5†). Hence, all FTIR analysis was done on liquid polysorbate 80 samples.
Interestingly, in dry state, with increasing concentrations of polysorbate 80, significant band shifts for –CH2 vibrations were observed from 2850 to 2866 cm−1 (for symmetric stretching vibrations) and from 2916 to 2926 cm−1 (for asymmetric stretching vibrations) (Fig. S1B†). These shifts represent the restriction in fluidity of acyl chain that may occur due to changes in concentration, temperature and solvent.40–43 We believe that in dry state, polysorbate 80 molecules are surrounded by very little or no water molecules. Thus in relatively hydrophobic environment, the acyl chain becomes more rigid. Similar behavior was reported for cetyltrimethylammonium bromide (CTAB) and cetylpyridinium chloride (CPC) with respect to change in their concentration in the aqueous solution as well as their physical form (dry or liquid state).44 In liquid state, no such shifts were observed (Fig. 1B) probably because of uniformity in acyl chain orientation and fluidity. Further, for dry state, in the region of 1800–1700 cm−1, polysorbate 80 (in the concentration range of 0.3–2.5 ng) showed a peak at 1726 cm−1 with small shoulder peak at 1740 cm−1 (Fig. S1C†). Above 2.5 ng these two peaks fused to give a single broad peak at 1744 cm−1. These peaks arise due to the stretching vibrations of –CO group and indicates degree of its hydration.45 Peak shift from 1726 cm−1 to 1744 cm−1 suggest decrease in hydrogen bonding because of increase in concentration simultaneously with reduction in surrounding water molecules. In liquid state, we observed same fused peaks in all concentrations without any shift (Fig. 1C). This indicates that –CO group remains hydrated suggesting hydrogen bonding between surfactant and water molecules. Similarly, in dry state, according to scissoring vibrations of –CH2 chain (1500–1400 cm−1) (Fig. S1D†), two peaks at 1423 cm−1 and 1470 cm−1 remain distinct in the range of 0.3 ng to 2.5 ng and fuse above 2.5 ng. These peaks are indication of mixture of trans and gauche conformers that exist because of acyl chain rotation of polysorbate 80 molecule.40,43 As physical state of polysorbate 80 molecule changes, acyl chain becomes rigid by acquiring ordered structure that is represented by the fused, broad peak showing maxima at 1440 cm−1 with shoulder peaks at 1423 cm−1 and 1470 cm−1 (Fig. S1D†). In liquid state, for all concentrations, the peaks at 1479 cm−1 and 1441 cm−1 can be observed (Fig. 1D); however, no fusion was observed which reiterates the possibility of uniformity of acyl chain structure in presence of water molecules. To summarize this, liquid samples showed similar FTIR spectra without any significant shifts unlike dry samples of polysorbate 80. Hence, for qualitative and quantitative analysis as well as for real-time FTIR study, we have used liquid samples of polysorbate 80 for FTIR analysis.
Synthesis and coating of PLGA nanoparticles
It is reasonable to assume that due to amphipathic nature, polysorbate 80 adsorbs on the surface of PLGA and other biodegradable polymers. But, the adsorption pattern, type of the adsorption process and adsorption kinetics of polysorbate 80 on PLGA nanoparticles is not well understood. Further, it has been shown that coating of PLGA nanoparticles with polysorbate 80 can be achieved within 30 minutes to 24 hours.14,17,22,33 Nonetheless, its relation to the adsorption process is not reported. To deduce this, we selected ten different concentrations of polysorbate 80 (0.1–30 mg ml−1). The percentage accuracy data (Fig. S5†) for our method of polysorbate 80 detection, suggests that the method is prone to high variations towards the low concentrations. Thus, the concentration below 0.1 mg ml−1 was not chosen. The concentration above 30 mg ml−1 was not taken because the equilibrium phase reaches before 5 minutes (see the subsequent paragraphs). All chosen concentrations of polysorbate 80, upon mixing with nanoparticle suspension, showed the absence of settled nanoparticles and no change in the turbidity. It suggests that the nanoparticles are stable which was further confirmed with DLS method (Table S3†). For delivery across BBB, the nanoparticles containing bioactive molecules and coated with polysorbate 80 should possess size below 200 nm.46 DLS data shows that nanoprecipitation and coating with polysorbate 80 yielded nanoparticles with ∼150 nm size and uniform distribution (Table S3†). Before coating, the nanoparticles remain stable and show no sign of aggregation in aqueous phase owing to the residual polyvinyl alcohol (PVA) on their surface. No significant change was observed in particle size and particle distribution (PDI) (t-test, P-value > 0.05) during different intervals of polysorbate 80 coating process (Fig. 2A and B). The scanning electron microscopy images of representative samples showed spherical shaped nanoparticles with no appreciable change in surface morphology after polysorbate 80 coating (Fig. S6†). | ||
| Fig. 2 Change in (A) particle size (intensity weighted diameter), (B) PDI with respect to time over the course of adsorption. (C) The amount of PVA quantified using UV-visible spectrophotometry at each time point. | ||
Confirmation of polysorbate 80 coating using differential scanning calorimetry (DSC)
To confirm the coating, DSC analysis was performed on uncoated PLGA nanoparticles (Fig. 3A–C) and randomly selected three representative polysorbate 80 coated nanoparticle samples (Fig. 3D–L). In DSC thermogram, all samples (uncoated and polysorbate 80 coated) showed an endothermic peak of PLGA polymer at 47 ± 2 °C (Fig. 3, blue arrows in A, D, G, J). This is glass transition temperature of PLGA and our results meet the criterion as per specifications (Tg = 42–47 °C) provided by the polymer manufacturer. Further, among selected coated nanoparticle samples, a multiple small peaks in the range of 64–68 °C were observed (Fig. 3, black arrows in H and K) which represent melting of polyethylene oxide (PEO) chains of polysorbate 80. Another peak was observed at 126 ± 2 °C in all concentrations (Fig. 3, red arrows in F, I, L) that represents the flash point of polysorbate 80.47,48 Multiples peaks in the range of 64–68 °C and 125–135 °C confirm presence of polysorbate 80 on PLGA nanoparticles. | ||
| Fig. 3 DSC thermograms of uncoated and three representative polysorbate 80 coated PLGA nanoparticle samples. | ||
Extraction of polysorbate 80 from PLGA nanoparticles
To quantify polysorbate 80 on PLGA nanoparticles, different strategies were tried. UV-visible spectrophotometric method was tested, but due to the interference from PLGA and PVA during the assay, it was not employed. Alternatively, FTIR analysis of polysorbate 80 coated nanoparticles was performed to observe the IR signatures of polysorbate 80 in primary absorption and second derivative spectra. PLGA showed interfering peaks in the region of acyl chain –CH2 band (Fig. S7†) of polysorbate 80. To resolve these problems, an extraction strategy was applied to separate polysorbate 80 from the nanoparticles. First, ethanol was used for the extraction.49,50 But, it led to the removal of PVA along with polysorbate 80, the interference of which was confirmed by FTIR (data not shown).Consequently, we developed a new method of extraction based on sequential dissolution, precipitation and centrifugation steps (Scheme 1). The hydrophobic PLGA polymer is soluble in various organic solvents like acetone, acetonitrile, dimethylformamide in which PVA remains insoluble.34,51,52 Taking advantage of differential solubility of two polymeric phases, PVA and PLGA were separated in subsequent steps as described in Scheme 1. At the end, the surfactant present in aqueous solution was analyzed with FTIR. Qualitatively, the identity of surfactant was assessed by comparing the IR spectrum of extracted polysorbate 80 with that of standard solution (Fig. 4). Use of different solvents and multiple centrifugation steps may degrade PLGA to lactic acid and glycolic acid. From the FTIR spectral comparison between extracted polysorbate 80, lactic acid and glycolic acid, this possibility was ruled out (Fig. 4, 5 and S4†). Complete separation of residual PVA was confirmed by UV spectrophotometry method53 and FTIR method. Significant difference was not obtained in PVA amount extracted from both coated and uncoated nanoparticles by UV spectrophotometric method (Fig. 2C). The IR spectrum of extracted polysorbate 80 was also devoid of PVA and PLGA peaks (Fig. 4, 5 and S4†).
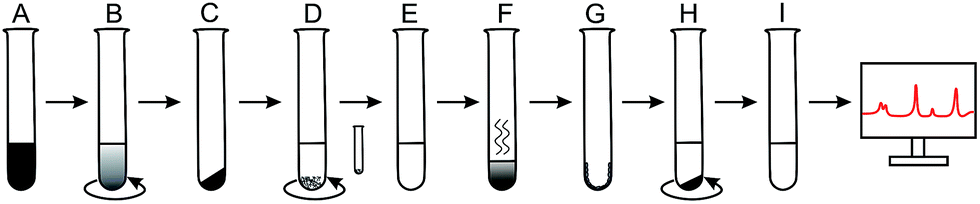 | ||
| Scheme 1 The process of extraction of polysorbate 80 from the nanoparticle surface. (A) Suspension of the coated nanoparticles collected at respective time points, (B) centrifugation steps to remove excess polysorbate 80 and to get pellet of washed nanoparticles, (C) dried pellet after removal of the supernatant, (D) addition of acetonitrile to dissolve the pellet. PVA precipitates out which can be removed out by centrifugation (small test tube), (E) clear supernatant containing PLGA and polysorbate 80 in acetonitrile, (F) evaporation of acetonitrile using vacuum desiccator, (G) PLGA and polysorbate 80 remained on the wall of test tube, (H) addition of water in which polysorbate 80 was solubilized but PLGA remains at the bottom of test tube. Finally ultracentrifugation at high speed to settle down PLGA precipitate, (I) clear aqueous supernatant containing polysorbate 80 and internal standard for FTIR analysis. | ||
 | ||
| Fig. 4 Spectral comparison of polysorbate 80 after extraction from nanoparticles with standard solution in the regions of stretching vibrations of acyl chain (A), stretching vibrations of ester group (B) and scissoring vibrations of acyl chain (C). | ||
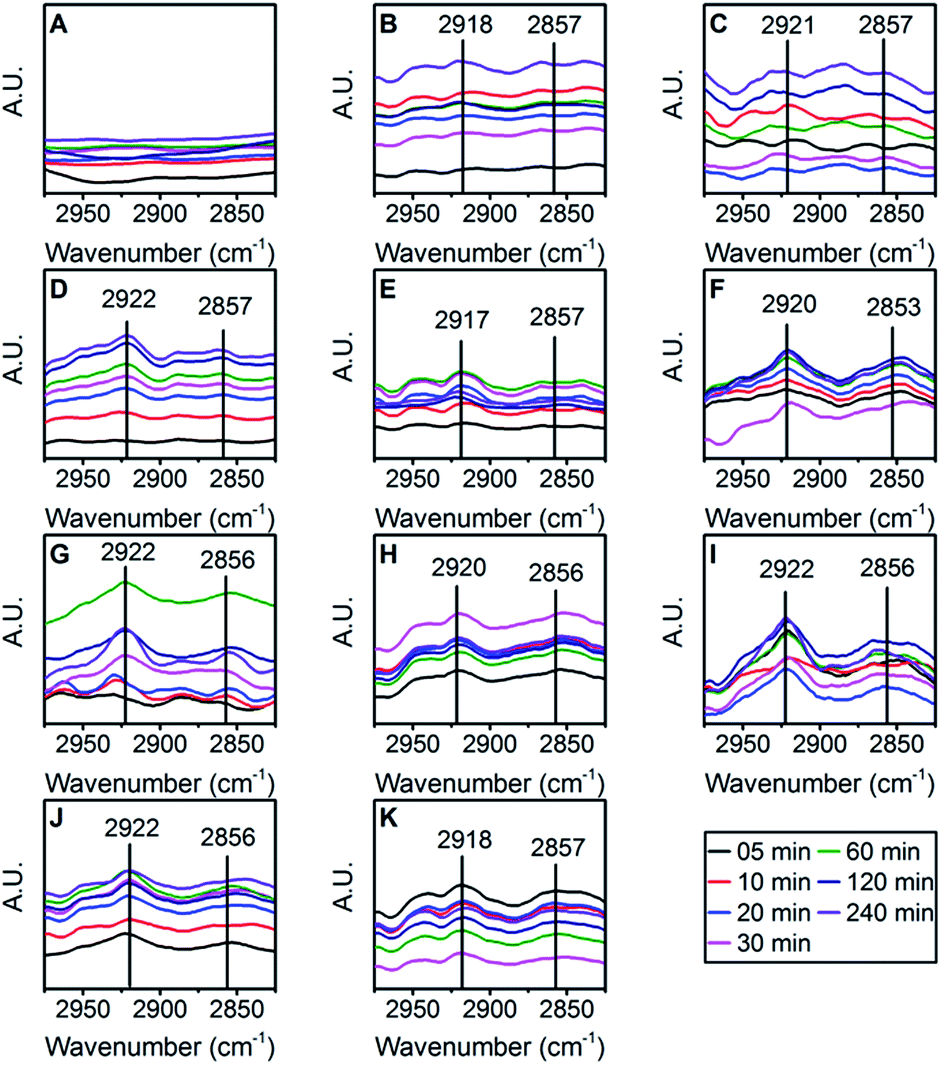 | ||
| Fig. 5 Acyl chain (–CH2) bands used for quantifying the extracted surfactant at each time point of the experiment started with 0.1 mg ml−1 (B), 0.3 mg ml−1 (C), 0.5 mg ml−1 (D), 1 mg ml−1 (E), 3 mg ml−1 (F), 5 mg ml−1 (G), 7.5 mg ml−1 (H), 10 mg ml−1 (I), 20 mg ml−1 (J) and 30 mg ml−1 (K) polysorbate 80. Control group (uncoated samples) show no bands in same region (A). | ||
Adsorption kinetics of polysorbate 80 on PLGA nanoparticles
To determine the kinetics of polysorbate 80 adsorption, PLGA nanoparticles were incubated with polysorbate 80 solution for 4 hours. Samples were collected at different time points, washed and polysorbate 80 was extracted to analyze by FTIR method for quantification. As stated previously, FTIR analysis of liquid samples yielded highly reproducible data with low standard error. Thus, for further analysis and calculations, we used the standard curve that was prepared after analyzing standard polysorbate 80 solutions in liquid state. Polysorbate 80 extracted from coated nanoparticles show acyl chain peak without any peak of PLGA and PVA, suggesting the accuracy of the extraction process (Fig. 4, 5 and S4†). The ratios of areas (AreaAcyl chain/AreaAzide) were fitted in the standard curve (Fig. 1A) to calculate the adsorbed amount of polysorbate 80. The amount obtained (Table S4†) was plotted against time to get the adsorption kinetic profile (Fig. 6A–J). The profile shows adsorption phase followed by an equilibration phase for concentration range within 0.1–5 mg ml−1. To identify the transition between adsorption and equilibration phases, t-test was used between consecutive time points of adsorption kinetics. For higher concentrations (7.5–30 mg ml−1), the exponential adsorption phase was masked because, as the starting concentration of polysorbate 80 increases, number of surfactant molecules in the vicinity of PLGA nanoparticles surface also increases. Under higher concentration gradient, it is possible that polysorbate 80 molecules move with higher rate of diffusion from bulk to nanoparticle surface.54 Thus the adsorption equilibrium time shifts towards lower values yielding inverse relationship with the starting concentration of polysorbate 80 (Fig. 7B). | ||
| Fig. 6 Adsorption kinetics of polysorbate 80 by ATR-method at the starting concentrations of 0.1–30 mg ml−1. The points indicate average value of adsorbed amount (μg mg−1) at each time point with standard deviation (n = 3) [note – all the lines were drawn using spline function as a guide for reader's eye to show the adsorption trend. The line does not necessarily represent actual average trend among repetitions of the experiment]. | ||
 | ||
| Fig. 7 Adsorption isotherm modeling: the adsorbed amount of polysorbate 80 (μg) per 1 mg of PLGA nanoparticles (A) showing exponential adsorption phase and saturation phase (A, inset) for selected concentrations of polysorbate 80. The time of adsorption equilibrium shows inverse relation to the starting concentration of polysorbate 80 (B). The adsorption data fitted in Langmuir model (C), Freundlich model (D), Temkin model (E) and Flory–Huggins model (F). | ||
Adsorption isotherm modeling
To determine the adsorption mechanism, Langmuir, Freundlich, Temkin and Flory–Huggins models were used55,56 (Fig. 7C–F). For all the concentrations, Langmuir model shows best fit with highest r2 value (0.95) (Fig. 7C) (Table S5†). It indicates that, polysorbate 80 forms the monolayer on the surface of PLGA nanoparticles. Also, the graph of Qeversus Ce (Fig. 7A) shows exponential rise at very low concentrations reaching to plateau phase which is classical feature of Langmuir adsorption process.57 Further, the value of separation factor (RL) calculated from this isotherm indicates that the surfactant follows ‘favorable adsorption process’ within given concentration range (calculation SC2 and Table S5†). The adsorption data at low concentrations showed fairly good fit to Flory–Huggins model (r2 = 0.91) (Fig. 7F) suggesting existence of intermolecular interactions55,58 between PLGA nanoparticles and polysorbate 80 molecules. Interestingly, the heat of adsorption (ΔG) obtained from this model (38 kJ Mol−1) suggests that the adsorption process is physisorption (Table S5†). Our data shows poor fit to Temkin model (Fig. 7E) suggesting absence of repulsive interactions and heterogeneity in adsorption process.56 Monolayer formation by polysorbate 80 was further confirmed by the least fit of Freundlich model (Fig. 7D). Overall analysis hints that polysorbate 80 adsorption leads to the monolayer formation by establishing non-covalent interactions with the nanoparticle surface. The adsorbed amount on nanoparticles for the concentrations above 0.3 mg ml−1 remains statistically same (10 ± 1 μg mg−1) (t-test, P-value > 0.05) suggesting the saturation of nanoparticle surface (Fig. 7A).Adsorption kinetic modeling
The adsorption kinetic profiles obtained from different starting concentrations of polysorbate 80 were analyzed for understanding the adsorption process and to support the conclusions drawn from isotherm modeling.59 Adsorption kinetic models describe the process of transfer of molecules from bulk solution to the solid surface. These models are classified into two categories – adsorption reaction models and adsorption diffusion models, each describing a different kinetic process.59 Among adsorption reaction models, we used Elovich model as the negative control, which originally describes chemisorption process, and found that it does not fit (r2 values ranging from 0.11–0.73) (Fig. S8†) to the adsorption kinetics data. This validates the physisorption of polysorbate 80 on PLGA nanoparticles.Further, from adsorption diffusion models, we used Weber–Morris model (Fig. S9†) which showed multi-linear adsorption kinetic profiles. Among selected concentration range of polysorbate 80, for lower concentrations (0.1–0.5 mg ml−1), we observed three linear regions in adsorption kinetics. On the other hand, rest all adsorption kinetics data showed only two linear regions (r2 values ranging from 0.90–0.97). This hints towards possibility of multistep adsorption process of polysorbate 80 molecules.
The inverse relation of time with adsorption equilibrium can be further supported by absence of multiple linear regions in kinetics data at higher concentrations. Fast movement of polysorbate 80 molecules from bulk towards nanoparticles limits us to capture exact kinetics and masks probable linear regions (if there are any). We propose that, a highly time sensitive technique which is capable of capturing real time data in micro/nanoseconds could possibly reveal exact linear regions in polysorbate 80 adsorption process.
From our observations of adsorption kinetic modeling, we speculate that polysorbate 80 adsorption is a multistep process comprised of initial diffusion from bulk to nanoparticle surface, diffusion through boundary layer, establishing interactions with PLGA surface and finally adsorption on nanoparticle surface59 (please see method SM5b† for detail description).
X-ray photoelectron spectroscopy (XPS) for surface characterization
XPS analysis provides a good measure for determination of presence and/or conformation of molecules adsorbed on a surface.60–62 In our experiments, broad scans of the lyophilized uncoated and polysorbate 80 coated nanoparticles showed presence of carbon (C, 1s) and oxygen (O, 1s) elements in all samples.Narrow scans for carbon element of uncoated and polysorbate 80 coated nanoparticles showed ensemble of 3 merged peaks in the range of 282–292 eV (Fig. S10,† part A and B). For all the samples four carbon functionalities were fixed for deconvolution as showed in Table S8.† Using binding energies (BE) associated with these functionalities, peak fitting was carried out (Fig. S10,† part A and B). In order to assess correctness of peak fitting, chi-square value and Abbe parameter were used. Chi-square value represents correlation between experimental data and fitted data. On the other hand, Abbe parameter dictates the distribution of residuals of fit. More accurate peak fitting is characterized by lower chi-square value. Abbe parameter takes value of 0 (poor fit, poorly distributed residuals) or 1 (good fit, statistically well distributed residuals) or 2 (poor fit, anti-correlated and oppositely distributed residuals). From the results of peak fitting experiment, chi-square value and Abbe parameter were found to be in the range of 2 ± 0.3 and 0.7 ± 0.1 respectively. Lower chi-square values and Abbe parameter close to unity confirmed correct peak fitting.
Further, binding energies of respective fitted peaks were plotted against starting concentration of polysorbate 80 and no shift was observed in any peak (Fig. S10,† part B, panel D). Shift in BE of a peak in XPS spectra denotes change in oxidation state of an element. Such shifts can be observed due to change in surrounding chemical environment and/or covalent bonding.63 However, no BE shift in XPS spectra confirmed absence of any chemical reaction and/or oxidation in adsorption process. This further affirms physisorption process of polysorbate 80 on PLGA nanoparticles. Deconvolution also yielded probable atomic% values of 4 functionalities. From the plot of atomic% versus polysorbate concentration, it can be concluded that the carbon% (BE = 284 eV) increased from ∼35% to ∼45% because of coating (Fig. S10,† part B, panel E). As compared to uncoated nanoparticles, the –CO% (ethoxy and ester group of polysorbate 80, BE = 286 eV) increased significantly. Similarly –COOH% (carbon of carboxylic group, BE = 288 eV) decreased. Both these observations suggest masking of lactide and glycolide groups and/or their possible engagement in non-covalent interactions with polysorbate 80 molecules. Overall XPS analysis confirms surface coverage and physisorption by polysorbate 80 molecules.
Real-time ATR-FTIR analysis of polysorbate 80 adsorption on PLGA nanoparticles
FTIR has also been used for determination of the conformational changes in various molecules like non-ionic surfactants, ionic surfactants, amphiphilic lipids which contain hydrophilic and hydrophobic moieties.40–43,45,64,65 These molecules change their conformation depending on various factors like temperature, concentration and the presence of other molecules in the vicinity. Considering three results of our experiments; (i) occurrence of band shifts due to concentration dependent acyl chain fluidity in dry state (Fig. S1B–D†), (ii) changes in overall spectra depending on physical state of polysorbate 80 molecule (either dry or liquid state) and (iii) existence of polysorbate 80-PLGA interactions (Flory–Huggins model, Fig. 7F), we hypothesized that, polysorbate 80 may show some changes in FTIR spectra in the presence of PLGA nanoparticles. Interestingly, after mixing polysorbate 80 solution with PLGA nanoparticle suspension on ZnSe crystal and real-time monitoring of this mixture, we observed significant band shifts for both hydrophilic (ester group) as well as hydrophobic (acyl chain of oleic acid) parts of polysorbate 80 (Fig. 8i, ii and S11†). In general, acyl chain packing in the lipids and lipid containing surfactants has been categorized based on three dimensional arrangement of –CH2 groups in three different classes namely; orthorhombic perpendicular, triclinic parallel and hexagonal packing. Orthorhombic perpendicular and triclinic parallel classes represent highly ordered and tightly packed acyl chain structure whereas, hexagonal packing shows disordered structure with more mobility and flexibility.40,66,67 These three classes show the signature bands in FTIR spectra.40 In our experiments, polysorbate 80 showed significant shift in symmetric stretching vibrations of acyl chain (from 2871 cm−1 to 2880 cm−1) (Fig. 8i and ii, panel A–D). Shift towards higher wavenumber in this band indicates that the structure becomes more flexible and disordered.40,43 As stated earlier, similar shift was observed in symmetric stretching vibration in different concentrations of polysorbate 80 when analyzed in dry state (Fig. S1B†). But, the magnitude of band shift in presence of PLGA nanoparticles (∼9 cm−1) is lesser as compared to that observed in different concentrations (∼8–16 cm−1). We speculate that different ratios of trans and gauche conformers are responsible for this change and it can be further explained by scissoring vibrations of the acyl chain.45 In presence of PLGA nanoparticles, the scissoring vibrations occur in the region of 1500–1400 cm−1 with the band shift towards lower wavenumber (from 1480 cm−1 to 1466 cm−1) (Fig. 8i and ii, panel I–L). It suggests that the acyl chain changes its ordered conformation to highly flexible hexagonal conformation with higher gauche/trans ratio.41 The ester group (–CO) of polysorbate 80 and its hydrogen bonding with neighboring molecules can be detected in the region of 1800–1700 cm−1. We observed significant shift in this band (from 1728 cm−1 to 1740 cm−1) (Fig. 8i and ii, panel E–H) suggesting direct interaction of this group with PLGA nanoparticle surface. Shift towards higher wavenumber indicates decrease in hydration of ester group.45 Interestingly, at lower concentrations of polysorbate 80 (0.5 and 1 mg ml−1), shifts were observed only in stretching and scissoring vibrations of acyl chain but not in vibrations of ester group (Fig. S11†). It suggests sensitivity of acyl chain towards PLGA nanoparticles surface during adsorption. From all these results, we speculate that as polysorbate 80 molecule approaches PLGA nanoparticle surface, the hydrophobic interactions dominates hydrogen bonding, leading to the adsorption and the monolayer formation.
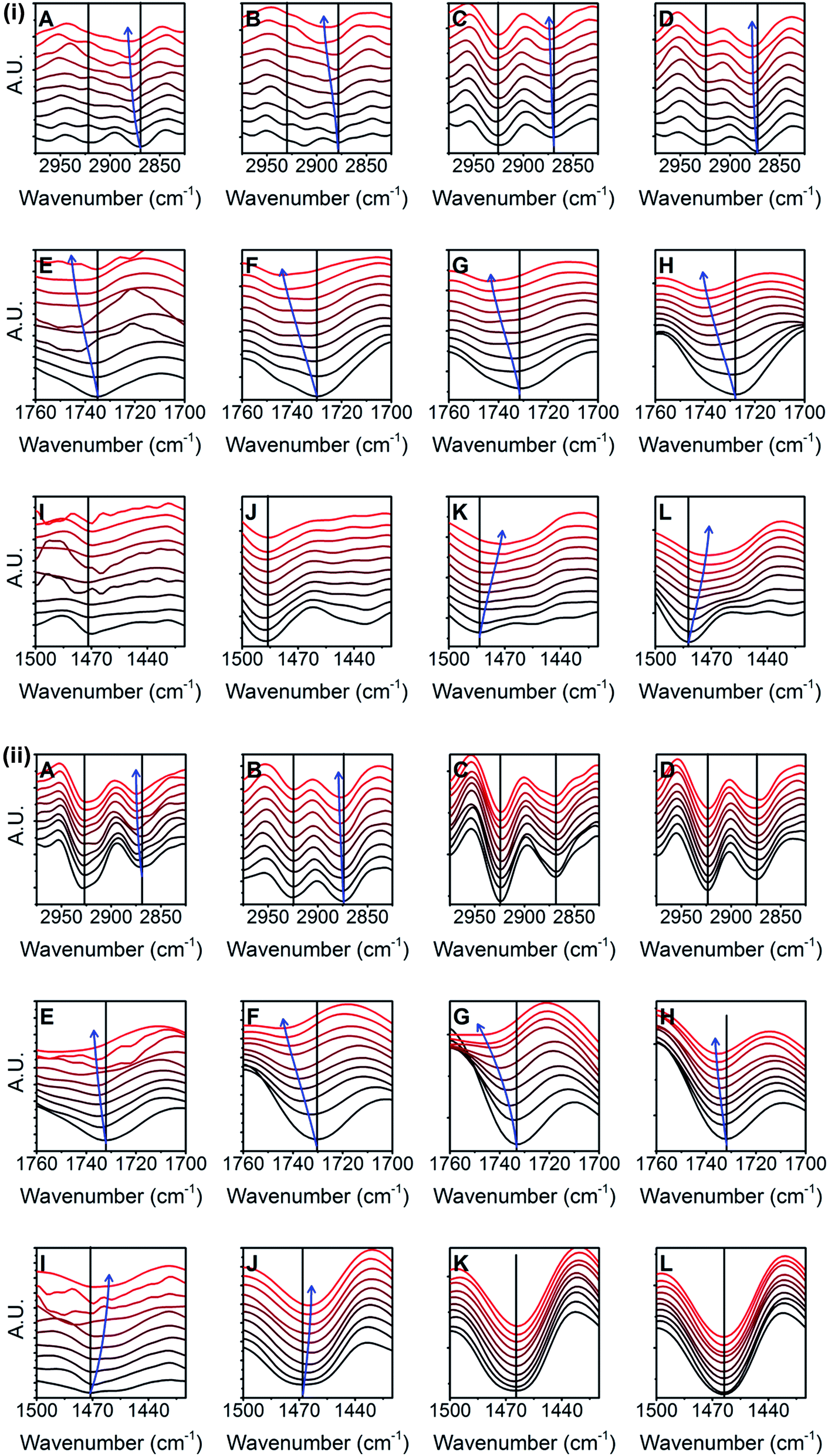 | ||
| Fig. 8 (i) Real-time FTIR data obtained for polysorbate 80 in presence of PLGA nanoparticles shows shifts in stretching vibrations of acyl chain (A–D), ester –CO group stretching vibrations (E–H), and scissoring vibrations of acyl chain (I–L). Each graph in 1st, 2nd, 3rd and 4th column represents experiment started with 0.5 mg ml−1, 1 mg ml−1, 3 mg ml−1 and 5 mg ml−1, respectively. (ii) Real-time FTIR data obtained for polysorbate 80 in presence of PLGA nanoparticles shows shifts in stretching vibrations of acyl chain (A–D), ester –CO group stretching vibrations (E–H), and scissoring vibrations of acyl chain (I–L). Each graph in 1st, 2nd, 3rd and 4th column represents experiment started with 7.5 mg ml−1, 10 mg ml−1, 20 mg ml−1 and 30 mg ml−1, respectively. | ||
On the other hand, polysorbate 80 alone (in absence of nanoparticles) shows no band shifts in acyl chain stretching vibrations (Fig. S12,† part A and B). In second derivative IR spectra, the band minima for these vibrations lie towards lower wavenumbers indicating ordered structure of acyl chain (Fig. S12,† part A and B, panel A–D). Further, no band shift was observed in the region of 1500–1400 cm−1 (Fig. S12,† part A and B, panel I–L). Ester group shows band at ∼1728 cm−1 (Fig. S12,† part A and B, panel E–H) suggesting increased hydration of polysorbate 80 molecule which is exactly opposite to its state in presence of PLGA nanoparticles. Overall, we conclude that polysorbate 80 loses its ordered structure, acquires highly flexible structure and decreases hydrogen bonding in the presence of PLGA nanoparticles during adsorption process. In presence of PLGA nanoparticles, at high concentration of polysorbate 80 (20 mg ml−1 and 30 mg ml−1) significant shifts were not observed because high number of stable micelles in the surrounding medium may suppress noticeable conformational change. Also, the resolution (4 cm−1) in our FITR experiments limits the detector to differentiate minor shifts in signature bands (<4 cm−1). Previously, using ellipsometry, it has been shown that on the hydrophobic surface of octadecyltriethoxysilane (OTE) polysorbate 80 forms a layer of 2 nm thickness. They attributed presence of one unsaturated double bond (in oleic acid moiety) to such peculiar behavior. They proposed that due to unsaturation and geometrical restriction, the molecule acquires the conformation parallel to the surface and lies flat on the surface.68 According to the results of our FTIR experiments, polysorbate 80 forms the monolayer with the involvement of hydrophobic and hydrophilic parts during adsorption on PLGA nanoparticles. While adsorption, it assumes flexible and disordered structure. Additionally, DLS results show no significant change in particle size after adsorption. Therefore, we speculate that polysorbate 80 may acquire flat structure on PLGA nanoparticle surface.
Conclusion
To summarize, we have synthesized polysorbate 80 coated PLGA nanoparticles and the coating was confirmed using ATR-FTIR, DSC and XPS methods. Further, we have developed a novel solvent extraction method for the retrieval of polysorbate 80 from nanoparticle surface and confirmed its complete extraction by ATR-FTIR method. We ensured no interference of other polymeric phases and confirmed the specificity of extraction method. Further, the kinetic profile of its adsorption on PLGA nanoparticles was determined using ATR-FTIR. The adsorption isotherm modeling suggests that polysorbate 80 follows physical adsorption process leading to the monolayer formation and develops interactions with PLGA nanoparticles under the given set of experimental conditions. Using adsorption kinetic models, we speculate that, polysorbate 80 molecules diffuse from bulk liquid towards nanoparticle surface and during equilibrium phase the nanoparticle surface becomes saturated with the surfactant. The time required for the adsorption equilibration is inversely proportional to the initial concentration of surfactant. At equilibration, 10 ± 1 μg polysorbate 80 gets adsorbed on unit mg of PLGA nanoparticles. The adsorption process has no significant impact on size, distribution and stability of nanoparticles. Using real-time FTIR, we confirmed the involvement of acyl chain (–CH2) and ester group (–CO) in the interactions of polysorbate 80 with PLGA nanoparticles. Real-time FTIR enabled us to deduce possible structural changes that polysorbate 80 underwent during initial phase of adsorption process. During adsorption, acyl chain acquires highly flexible structure and the hydration level of ester group decreases suggesting possibility of increased hydrophobic interactions. Thus by judging FTIR and DLS results, we propose that polysorbate 80 acquires flat structure on nanoparticle surface. Understanding such mechanistic details of adsorption of a surfactant on the nanoparticle surface is important in the field of nanomedicine, advanced drug delivery systems and pharmacology. It can be useful for studying nanoparticle interactions with key cellular players, for example, protein–nanoparticle interactions, which ultimately decide their in vivo fate. Studying the mechanistic details of adsorption and quantification of polysorbate 80 will be helpful in designing the optimized drug delivery system with specific organ targeting and improved biological half-life.
Experimental
Materials
PLGA 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (acid terminated, avg. mol. wt 7000–16000), poly(vinyl) alcohol (PVA; mol. wt 30000–70000 Da, 87–90% hydrolyzed), potassium iodide were purchased from Sigma-Aldrich, Delhi, India. Acetonitrile, dimethyl sulfoxide (DMSO) and iodine (resublimed) were procured from Merck, Mumbai, India. Polysorbate 80 (polyoxyethylene sorbitan mono-oleate or Tween-80®), sodium azide and boric acid were obtained from HiMedia laboratories, Mumbai, India. For all experiments, the water obtained from MilliQ system (Millipore unit, 18.2 Mohm cm at 25 °C) was used.
:50 (acid terminated, avg. mol. wt 7000–16000), poly(vinyl) alcohol (PVA; mol. wt 30000–70000 Da, 87–90% hydrolyzed), potassium iodide were purchased from Sigma-Aldrich, Delhi, India. Acetonitrile, dimethyl sulfoxide (DMSO) and iodine (resublimed) were procured from Merck, Mumbai, India. Polysorbate 80 (polyoxyethylene sorbitan mono-oleate or Tween-80®), sodium azide and boric acid were obtained from HiMedia laboratories, Mumbai, India. For all experiments, the water obtained from MilliQ system (Millipore unit, 18.2 Mohm cm at 25 °C) was used.
Methods
000g for 1 hour (Sorvall microultracentrifuge, Thermo Scientific Ltd., Rotor S-55-S) to remove excess PVA. The nanoparticles were washed once, resuspended in 4 ml water and used for further experiments.
000g during all these steps. Acetonitrile, which contains PLGA polymer and polysorbate 80 was then evaporated using vacuum desiccator for about 2 hours (Scheme 1, steps E–G). After drying 150 μl water was added to dissolve polysorbate 80. PLGA remains insoluble and can be observed in the form of white particles at the bottom of tube. PLGA was then separated by ultracentrifugation at 150000g for 2 hours (Sorvall microultracentrifuge, Thermo Scientific Ltd., Rotor S80-AT2) (Scheme 1, step H). Ultracentrifugation step is necessary because very minute amount of PLGA, if present any, will create sharp interfering FTIR signal. To 50 μl of clear supernatant, 1 μl internal standard (1 mg ml−1 sodium azide) was added and FTIR analysis was performed to characterize polysorbate 80 (Scheme 1, step I).
For the data files obtained from real-time FTIR analysis (repeated measurement mode), smoothening and baseline correction was done as mentioned above. After baseline correction, second derivative was calculated using Origin Pro software (64 bit, Sr3, b275, OriginLab Corporation, Northampton, MA01060, USA) and presented in stacking format to highlight the band shifting.
Conflict of interest
The authors would like to declare conflict of interest according to the provisional patent application number 3003/DEL/2015.Acknowledgements
A. K. T. sincerely thanks Department of Biotechnology, Government of India, for project funding. A. S. J. and A. Y. G. sincerely thank Indian Institute of Technology (IIT) Kanpur for senior research fellowship. We are grateful to Dr Dhirendra S. Katti, IIT Kanpur for allowing multi-well plate reader facility. We are thankful to Advanced Imaging Center of IIT Kanpur for providing electron microscopy facility. A. S. J. also wants to thank Mr Kamlesh (Advanced Center for Material Science (ACMS) department, IIT Kanpur) for helping and allowing us to do DSC and XPS study. A. S. J. and A. Y. G. thank Mr Suhail Rizvi and Mr Vishesh Sood for critical discussion about ‘Adsorption and adsorption isotherm analysis’.Notes and references
- T. F. Tadros, Applied Surfactants: Principles and Applications, Wiley-VCH Verlag GmBH & Co. KGaA, Germany, 2005 Search PubMed.
- M. R. Vidal-Paruta and L. D. King, J. Pharm. Sci., 1964, 53, 1217–1220 CrossRef CAS PubMed.
- A. Patist, S. S. Bhagwat, K. W. Penfield, P. Aikens and D. O. Shah, J. Surfactants Deterg., 2000, 3, 53–58 CrossRef CAS.
- A. C. Braun, D. Ilko, B. Merget, H. Gieseler, O. Germershaus, U. Holzgrabe and L. Meinel, Eur. J. Pharm. Biopharm., 2015, 94, 559–568 CrossRef CAS PubMed.
- D. A. El-Setouhy, E. B. Basalious and N. S. Abdelmalak, Eur. J. Pharm. Biopharm., 2015, 94, 386–392 CrossRef CAS PubMed.
- D. Desai, B. Wong, Y. Huang, Q. Ye, D. Tang, H. Guo, M. Huang and P. Timmins, J. Pharm. Sci., 2014, 103, 920–926 CrossRef CAS PubMed.
- Y. Lu, Y.-Y. Wang, N. Yang, D. Zhang, F.-Y. Zhang, H.-T. Gao, W.-T. Rong, S.-Q. Yu and Q. Xu, Toxicol. Sci., 2014, 139, 317–327 CrossRef CAS PubMed.
- B. S. Chang, B. S. Kendrick and J. F. Carpenter, J. Pharm. Sci., 1996, 85, 1325–1330 CrossRef CAS PubMed.
- S. Ding, J. Pharm. Biomed. Anal., 1993, 11, 95–101 CrossRef CAS PubMed.
- Y. Chen and L. Liu, Adv. Drug Delivery Rev., 2012, 64, 640–665 CrossRef CAS PubMed.
- J. Kreuter, P. Ramge, V. Petrov, S. Hamm, S. E. Gelperina, B. Engelhardt, R. Alyautdin, H. von Briesen and D. J. Begley, Pharm. Res., 2003, 20, 409–416 CrossRef CAS.
- M. Masserini, ISRN Biochem., 2013, 1–18 CrossRef PubMed.
- J.-C. Olivier, NeuroRx, 2005, 2, 108–119 CrossRef PubMed.
- W. Sun, C. Xie, H. Wang and Y. Hu, Biomaterials, 2004, 25, 3065–3071 CrossRef CAS PubMed.
- T. M. Goppert and R. H. Muller, J. Drug Targeting, 2005, 13, 179–187 CrossRef PubMed.
- J. Kreuter, R. N. Alyautdin, D. A. Karkevich and B. A. Sabel, US Pat., 6117454 (A), 2000.
- B. Petri, A. Bootz, A. Khalansky, T. Hekmatara, R. Müller, R. Uhl, J. Kreuter and S. Gelperina, J. Controlled Release, 2007, 117, 51–58 CrossRef CAS PubMed.
- C. J. Cheng, G. T. Tietjen, J. K. Saucier-Sawyer and W. M. Saltzman, Nat. Rev. Drug Discovery, 2015, 14, 239–247 CrossRef CAS PubMed.
- S. Simsek, H. Eroglu, B. Kurum and K. Ulubayram, J. Microencapsulation, 2013, 30, 10–20 CrossRef CAS PubMed.
- Y. C. Chen, W. Y. Hsieh, W. F. Lee and D. T. Zeng, J. Biomater. Appl., 2013, 27, 909–922 CrossRef PubMed.
- S. Gelperina, O. Maksimenko, A. Khalansky, L. Vanchugova, E. Shipulo, K. Abbasova, R. Berdiev, S. Wohlfart, N. Chepurnova and J. Kreuter, Eur. J. Pharm. Biopharm., 2010, 74, 157–163 CrossRef CAS PubMed.
- M. Kolter, M. Ott, C. Hauer, I. Reimold and G. Fricker, J. Controlled Release, 2015, 197, 165–179 CrossRef CAS PubMed.
- G. Mittal, H. Carswell, R. Brett, S. Currie and M. N. V. R. Kumar, J. Controlled Release, 2011, 150, 220–228 CrossRef CAS PubMed.
- H. Wesemeyer, B. W. Müller and R. H. Müller, Int. J. Pharm., 1993, 89, 33–40 CrossRef CAS.
- H. Carstensen, B. W. Müller and R. H. Müller, Int. J. Pharm., 1991, 67, 29–37 CrossRef CAS.
- K. P. Sharma, V. K. Aswal and G. Kumaraswamy, J. Phys. Chem. B, 2010, 114, 10986–10994 CrossRef CAS PubMed.
- J. Kreuter, D. Shamenkov, V. Petrov, P. Ramge, K. Cychutek, C. Koch-Brandt and R. Alyautdin, J. Drug Targeting, 2002, 10, 317–325 CrossRef CAS PubMed.
- N. Watrous-Peltier, J. Uhl, V. Steel, L. Brophy and E. Merisko-Liversidge, Pharm. Res., 1992, 9, 1177–1183 CrossRef CAS.
- S. D. Tröster, U. Müller and J. Kreuter, Int. J. Pharm., 1990, 61, 85–100 CrossRef.
- C. M. Keck, M. Jansch and R. H. Müller, Pharmaceutics, 2013, 5, 36–68 CrossRef CAS PubMed.
- B. Gbaguidi, O. C. Germay and S. Lardau, US Pat., 20150129766 A1, 2015.
- J. P. R. Day, R. A. Campbell, O. P. Russell and C. D. Bain, J. Phys. Chem. C, 2007, 111, 8757–8774 CAS.
- R. M. Koffie, C. T. Farrar, L. J. Saidi, C. M. William, B. T. Hyman and T. L. Spires-Jones, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18837–18842 CrossRef CAS PubMed.
- A. S. Joshi and A. K. Thakur, J. Pept. Sci., 2014, 20, 630–639 CrossRef CAS PubMed.
- R. F. Tabor, J. Eastoe and P. Dowding, Langmuir, 2009, 25, 9785–9791 CrossRef CAS PubMed.
- R. T. M. Fraser, Anal. Chem., 1959, 31, 1602–1603 CrossRef CAS.
- C.-H. Kuo, D. Y. Vorobyev, J. Chen and R. M. Hochstrasser, J. Phys. Chem. B, 2007, 111, 14028–14033 CrossRef CAS PubMed.
- F. M. Mirabella, J. Polym. Sci., Polym. Phys. Ed., 1983, 21, 2403–2417 CrossRef CAS.
- J. M. Green, Anal. Chem., 1996, 68, 305A–309A CrossRef CAS.
- R. N. A. H. Lewis and R. N. McElhaney, Biochim. Biophys. Acta Biomembr., 2013, 1828, 2347–2358 CrossRef CAS PubMed.
- R. Mendelsohn and D. J. Moore, Chem. Phys. Lipids, 1998, 96, 141–157 CrossRef CAS PubMed.
- T. Kawai, H. Kamio, T. Kondo and K. Kon-No, J. Phys. Chem. B, 2005, 109, 4497–4500 CrossRef CAS PubMed.
- T. C. Wong, N. B. Wong and P. A. Tanner, J. Colloid Interface Sci., 1997, 186, 325–331 CrossRef CAS PubMed.
- K. H. S. Kung and K. F. Hayes, Langmuir, 1993, 9, 263–267 CrossRef CAS.
- A. Dicko, H. Bourque and M. Pézolet, Chem. Phys. Lipids, 1998, 96, 125–139 CrossRef CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 Search PubMed.
- K. Pramod, C. V. Suneesh, S. Shanavas, S. H. Ansari and J. Ali, J. Anal. Sci. Technol., 2015, 6, 1–14 Search PubMed.
- J.-Y. Fang, C.-L. Fang, C.-H. Liu and Y.-H. Su, Eur. J. Pharm. Biopharm., 2008, 70, 633–640 Search PubMed.
- L. Zhao, Y. Yu, L. Song, M. Ruan, X. Hu and A. Larbot, Appl. Catal., A, 2004, 263, 171–177 Search PubMed.
- R. van Grieken, G. Calleja, G. D. Stucky, J. A. Melero, R. A. García and J. Iglesias, Langmuir, 2003, 19, 3966–3973 Search PubMed.
- K. C. Song, H. S. Lee, I. Y. Choung, K. I. Cho, Y. Ahn and E. J. Choi, Colloids Surf., A, 2006, 276, 162–167 CrossRef CAS.
- M. I. Baker, S. P. Walsh, Z. Schwartz and B. D. Boyan, J. Biomed. Mater. Res., Part B, 2012, 100, 1451–1457 CrossRef PubMed.
- C. A. Pannuti, V. D. Lynch, A. J. Monte-Bovi and J. J. Sciarra, J. Pharm. Sci., 1966, 55, 535 CrossRef CAS.
- C.-R. Lee, H.-S. Kim, I.-H. Jang, J.-H. Im and N.-G. Park, ACS Appl. Mater. Interfaces, 2011, 3, 1953–1957 CAS.
- K. Y. Foo and B. H. Hameed, Chem. Eng. J., 2010, 156, 2–10 CrossRef CAS.
- C. J. Pursell, H. Hartshorn, T. Ward, B. D. Chandler and F. Boccuzzi, J. Phys. Chem. C, 2011, 115, 23880–23892 CAS.
- S. Louguet, A. C. Kumar, N. Guidolin, G. Sigaud, E. Duguet, S. Lecommandoux and C. Schatz, Langmuir, 2011, 27, 12891–12901 CrossRef CAS PubMed.
- B. Kronberg and P. Stenius, J. Colloid Interface Sci., 1984, 102, 410–417 CrossRef CAS.
- H. Qiu, L. Lv, B.-c. Pan, Q.-j. Zhang, W.-m. Zhang and Q.-x. Zhang, J. Zhejiang Univ., Sci., A, 2009, 10, 716–724 CrossRef CAS.
- P. D. Scholes, A. G. A. Coombes, L. Illum, S. S. Davis, J. F. Watts, C. Ustariz, M. Vert and M. C. Davies, J. Controlled Release, 1999, 59, 261–278 CrossRef CAS PubMed.
- G. Cappelletti, C. L. Bianchi and S. Ardizzone, Appl. Surf. Sci., 2006, 253, 519–524 CrossRef CAS.
- J. Xu, L. Shi and M. Ye, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2297–2302 CrossRef CAS.
- Y. Yoshida, B. Van Meerbeek, Y. Nakayama, J. Snauwaert, L. Hellemans, P. Lambrechts, G. Vanherle and K. Wakasa, J. Dent. Res., 2000, 79, 709–714 CrossRef CAS PubMed.
- B. Mädler, H. Binder and G. Klose, J. Colloid Interface Sci., 1998, 202, 124–138 CrossRef.
- F. G. Wu, J. J. Luo and Z. W. Yu, Langmuir, 2010, 26, 12777–12784 CrossRef CAS PubMed.
- D. M. Small, J. Lipid Res., 1984, 25, 1490–1500 CAS.
- D. L. Dorset, S. W. Hui and C. M. Storzewski, J. Supramol. Struct., 1976, 5, 1–14 CrossRef CAS PubMed.
- M. Graca, J. H. H. Bongaerts, J. R. Stokes and S. Granick, J. Colloid Interface Sci., 2007, 315, 662–670 CrossRef CAS PubMed.
- M. Pirzer and J. Sawatzki, US Pat., 7359815 B2, 2008.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods in detail, calculations, additional tables and figures. See DOI: 10.1039/c6ra07699h |
| This journal is © The Royal Society of Chemistry 2016 |