
Preparation and characterization of interface-modified PLA/starch/PCL ternary blends using PLLA/triclosan antibacterial nanoparticles for medical applications
Abstract
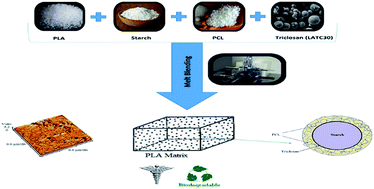
In this study, the interface-modified ternary blends based on polylactic acid/starch/polycaprolactone were prepared via melt blending. On one hand, the addition of triclosan-loaded polylactic acid (LATC30) nanoparticles imparted antibacterial properties to the blend, and on the other hand, the interfacial affinity between hydrophilic starch and hydrophobic polyesters was improved by establishing interactions between –Cl groups of triclosan and ester groups of PCL. AFM and rheological results demonstrated that the starch particles are very coarse in PLA/starch 50:50 (PLAS50) but as the content of PCL increased, the extent of chemical interactions between starch and PCL was increased which resulted in a much better dispersion and homogeneity of starch within the ternary blends. Tensile analysis indicated that the interface modification of starch by the hydrophobic PCL and triclosan significantly improved the elongation at break and tensile strength values of PLAS50, and the mechanical character of blends was transformed from brittle to ductile. Biodegradability was also investigated and the triclosan release rate was tuned, while the blends are also antibacterial and their cell-viability was approved. Overall, the ternary blending approach of PLA/starch/PCL using triclosan is a promising technique to extend the property range of PLA suitable for many medical applications.
 Please wait while we load your content...
Please wait while we load your content...

