
Graphene oxide: strategies for synthesis, reduction and frontier applications
Rajesh Kumar Singh
a,
Rajesh Kumar
*b and
Dinesh Pratap Singh
*c
aSchool of Physical & Material Sciences, Central University of Himachal Pradesh (CUHP), Kangra, Dharamshala, HP-176215, India
bCenter for Semiconductor Components and Nanotechnology (CCS Nano), University of Campinas (UNICAMP), 13083-870 Campinas, Sao Paulo, Brazil. E-mail: rajeshbhu1@gmail.com
cDepartamento de Física, Universidad de Santiago de Chile, Avenida Ecuador 3493, Estación Central, Santiago 9170124, Chile. E-mail: dineshpsingh@gmail.com
First published on 28th June 2016
Abstract
Till now, several innovative methods have been developed for the synthesis of graphene materials including mechanical exfoliation, epitaxial growth by chemical vapor deposition, chemical reduction of graphite oxide, liquid-phase exfoliation, arc discharge of graphite, in situ electron beam irradiation, epitaxial growth on SiC, thermal fusion, laser reduction of polymers sheets and unzipping of carbon nanotubes etc. Generally large scale graphene nanosheets are reliably synthesized utilizing other forms of graphene-based novel materials, including graphene oxide (GO), exfoliated graphite oxide (by thermal and microwave), and reduced graphene oxide. The degree of GO reduction and number of graphene layers are minimized mainly by applying two approaches via chemical or thermal treatments. The promising and excellent properties together with the ease of processability and chemical functionalization makes graphene based materials especially GO, ideal candidates for incorporation into a variety of advanced functional materials. Chemical functionalization of graphene can be easily achieved, by the introduction of various functional groups. These functional groups help to control and manipulate the graphene surfaces and help to tune the properties of the resulting hybrid materials. Importantly, graphene and its derivatives GO, have been explored in a wide range of applications, such as energy generation/storage, optical devices, electronic and photonic devices, drug delivery, clean energy, and chemical/bio sensors. In this review article, we have incorporated a general introduction of GO, its synthesis, reduction and some selected frontier applications.
 Rajesh Kumar Singh | Rajesh Kumar Singh is an Asst. Professor in the School of Physical and Material Sciences, Central University of Himachal Pradesh, India. He obtained his Ph.D. (2011) degree in Physics from Institute of Sciences, Banaras Hindu University (B.H.U) Varanasi, India. He then spent 4 years (2011–2015) at the Indian Institute of Technology (B.H.U.) Varanasi as a postdoctoral fellow. His current research interests include hydrogen storage, solid oxide fuel cells, synthesis of carbon nanostructures-metal/metal oxide self-assembly and their applications in different areas. He has published one international book chapter, two review and several peer-reviewed international papers. |
 Rajesh Kumar | Rajesh Kumar is a Postdoctoral Fellow in the Center for Semiconductor Components and Nanotechnology, University of Campinas (UNICAMP), Sao Paulo, Brazil. He received his B.S and M.S. (Physics) degree from Allahabad University, India. He earned his Ph.D. (Physics) (2011) degree from Department of Physics, Banaras Hindu University (B.H.U), India. He has worked in KAIST, Daejeon (Postdoctoral Researcher) and Yonsei University (Research Professor), Seoul, South Korea. His main research interests are the synthesis, characterization, modifications, assemblies and applications of the graphitic carbon nano-materials as graphene and CNTs. He has published two international book chapters, two review articles and several research articles in peer-reviewed international journals. |
 Dinesh Pratap Singh | Dr. Dinesh Pratap Singh received M.Sc. (2000) and Ph.D. (2007) degree in Physics from the Banaras Hindu University (B.H.U), Varanasi India. After postdoctoral studies from Southern Illinois University Carbondale, Illinois, USA, he became an assistant professor of Physics at University of Santiago in Chile (2010). His current research interests include, graphene based solar cells, synthesis of nanomaterials, high temperature nanotribology and biomedical applications of nanomaterials. He has published several refereed journal articles, review articles and book chapters. |
1. Introduction
Graphene is an exciting material of the 21st century, which has a one-atom-thick 1D planar sheet of sp2-bonded carbon atoms in a honeycomb crystal lattice with exceptionally high crystal quality and electronic properties which has emerged as a rapidly rising star in the field of material science.1–5 It is also known as the mother element of carbon allotropes, including graphite, carbon nanotubes, and fullerenes.3,6 Its peculiar properties such as high intrinsic mobility,7,8 thermal conductivity,9 high theoretical specific surface area,10 high Young's modulus,11 high optical transmittance12 and good electrical conductivity13 merit attention for various novel potential applications. These properties were experimentally studied and verified in last several decades14–19 and various top-up and bottom-up synthesis approach have been developed for high quality graphene using micromechanically exfoliation of graphite layers,20 graphene grown on 6H–SiC,21 large area graphene grown on copper substrates22 etc.Nowadays, graphene can be produced by micro-mechanical exfoliation of highly ordered pyrolytic graphite,20 epitaxial growth,17,23,24 and chemical vapor deposition (CVD).25,26 These three methods can produce graphene with a relatively perfect structure and excellent properties. However in comparison, graphene produced by reduction of graphene oxide (GO) has some important characteristics as it is produced using graphite material by cost-effective chemical methods in large scale. It is also highly hydrophilic and forms stable aqueous colloids to facilitate the assembly of macroscopic structures by simple and cheap solution processes. As a result, GO and reduced GO (rGO) are still hot topics in the research area and development of graphene, especially in regard to mass applications.
Synthesis of graphite oxide by oxidation of graphite through various chemical routes is the fundamental need for graphene-based materials. GO and graphene are highly promising for various applications due to their unusual thermal, electrical and optical properties. GO produced from graphite using various chemical oxidation routes,27–30 has attracted considerable attention as a potential material for use in various industrial applications such as photovoltaic cells, capacitors, sensors, and transparent electrodes.31–35 One particular branch of graphene research deals with GO and graphene synthesized from graphite oxide. Graphite oxide is generally obtained by oxidation of natural graphite powder with various oxidants in acidic media.
The presence of several groups like epoxy, hydroxyl, carboxyl and carbonyl on GO are well recognised and accepted as major functionalities attached to it. Several structural models of graphite oxide comprising these groups have been reported so far in the literature.36–39 GO can be considered as a precursor for graphene synthesis by either chemical, thermal or microwave reduction processes. GO consists of a 1D single-layer of graphite oxide and is usually produced by the chemical treatment of graphite through oxidation.36,40 The GO mainly consists of individual sheets of graphene with attachments of oxygen functional groups on both the basal planes and edges.41 It has different structural models and contains various oxygen functional groups on its surfaces. The oxygen functional groups have been identified as hydroxyl and epoxy groups present on the basal plane, carboxy, carbonyl, and phenol groups at the edge of GO sheets.42–44 For the synthesis of GO, we first need to synthesize graphite oxide by different chemical process route.
2. Synthesis of graphene/graphite oxide: oxidation of bulk graphite powder
From last several years, both theoretically and experimentally, considerable effort has been directed toward understanding the structure of graphite oxide. The GO is currently the most common precursor used for the synthesis of graphene materials. The focused study of the GO structure is derived from the structural analysis of graphite oxide itself. Its can be prepared by the intercalation and oxidation of graphite powder. For synthesis of GO in bulk amount, we first need to synthesize graphite oxide applying any of the methods discussed below. The history of GO is very interesting especially when we deal with its synthesis. The first report on the synthesis of GO by the intercalation of graphite sheets was given by a Schafhacutl in 1840. Schafhacutl for the very first time attempted to exfoliate graphite, when he tried to purify the impure graphite “kish” from iron smelters.45–47The well known methods used for the synthesis of graphite oxide are Brodie method,27 Staudenmaier method,29 Hofmann method30,48 and Hummers method28 and also their modified and improved forms. In these methods, initially graphite powder is chemically reacted with acids (HCl, H2SO4 and HNO3 etc.) followed by the intercalation of alkali metals (alkali metal compounds KClO3, KMnO4, NaNO3 etc.) into the graphitic layers which further helps in the breaking of graphitic layers into small pieces.
2.1. Brodie's oxidation method
The oxidation of graphite in the presence of potassium chlorate (KClO3) and fuming nitric acid was developed by B.C. Brodie in 1859. He was the pioneering one who treated graphitic powder with KClO3 in concentrated fuming HNO3 (ref. 27) and got a new compound which later on was determined to contain carbon, oxygen and hydrogen resulting in an increase in the overall mass of the flake graphite. He isolated the crystals of the material, but the interfacial angles of the crystal lattice were unable to be measured via reflective goniometry. Successive oxidative treatments resulted in a further increase in the oxygen content, reaching a limit after several reactions. The C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H:O percentage composition was determined to be 61.04:1.85:37.11. According to his elemental analysis, the molecular formula for the final product was C11H4O5. He found that the as synthesized material are dispersible in basic or pure water, but not in acidic media, which prompted him to term the material as graphic acid. After heating (220 °C), the C:H:O composition of this material changed to 80.13:0.58:19.29, with a loss of carbonic acid and carbonic oxide. This method has several flaws including long reaction time as well as release of toxic gases during reaction. Although some research groups used this method for the production of graphite oxide and cited in their published literature.36,49–54
:H:O percentage composition was determined to be 61.04:1.85:37.11. According to his elemental analysis, the molecular formula for the final product was C11H4O5. He found that the as synthesized material are dispersible in basic or pure water, but not in acidic media, which prompted him to term the material as graphic acid. After heating (220 °C), the C:H:O composition of this material changed to 80.13:0.58:19.29, with a loss of carbonic acid and carbonic oxide. This method has several flaws including long reaction time as well as release of toxic gases during reaction. Although some research groups used this method for the production of graphite oxide and cited in their published literature.36,49–54
2.2. Staudenmaier method: fuming HNO3, concentrated H2SO4 acid and KClO3
One of the improvements on Brodie's work happened in 1898 by L. Staudenmaier using excess of the oxidizing agent and concentrated sulfuric acid as extra additive.29 He improves the Brodie's KClO3-fuming HNO3 preparation by (i) adding multiple aliquots of potassium chlorate solution into the reaction mixture over the course of reaction and adding concentrated sulfuric acid (H2SO4) to increase the acidity of the mixture. These changes led to a highly oxidized graphite oxide in a single reaction vessel, thus simplified the GO synthesis process. This slight change in the synthesis procedure resulted in an overall extent of oxidation similar to Brodie's multiple oxidation approach (C:O ∼ 2:1). However, this Staudenmaier's preparation method was time consuming and hazardous: the addition of potassium chlorate typically lasted over a week, and the chlorine dioxide evolved needed to be removed by an inert gas, while explosion was a constant hazard. Therefore, the further modification or development of the new process for oxidation was still under investigation. This method and its modified form has also been used by several other groups for the synthesis of graphite oxide.55–60
2.3. Hofmann method: concentrated HNO3 acid, concentrated H2SO4 acid and KClO3
In 1937, Hofmann et al.30,48 used concentrated sulfuric acid in combination with concentrated nitric acid and KClO3 for the oxidation of graphite for the preparation of graphite oxide. The KClO3 is a highly strong oxidizing agent and it oxidizes the graphite powder in acids solution and typically also is an in situ source of dioxygen, which acts as the reactive species. Several research groups have synthesized GO for different application purpose using Hofmann method.61,622.4. Hummers method: concentrated H2SO4 acid in the presence of KMnO4 and NaNO3
In 1958, Hummers and Offeman shows an alternate method for the oxidation of graphite by reacting graphite powder with a mixture of concentrated H2SO4, NaNO3 and KMnO4.28 First graphite powder (100 g) and sodium nitrate (50 g) were stirred in sulfuric acid (2.3 liters) and cooled to 0 °C in ice bath. After that potassium permanganate (300 g) was added to the suspension. During the stirring, the suspension becomes thick and was then further diluted using hot water and treated with H2O2 (3%) to reduce the residual permanganate and manganese dioxide to colorless soluble manganese sulfate. Finally, the diluted suspension was filtered and washed several time with warm water to remove the soluble salt of mellitic acid. The dry form of graphitic oxide was obtained by centrifugation followed by dehydration at 40 °C over phosphorus pentoxide in vacuum. In recent time, several research groups have used Hummers improved method for the synthesis of graphite oxide.10,63–71 All the well known synthesis methods for graphite oxidation using chemical routes have been summarized in Fig. 1.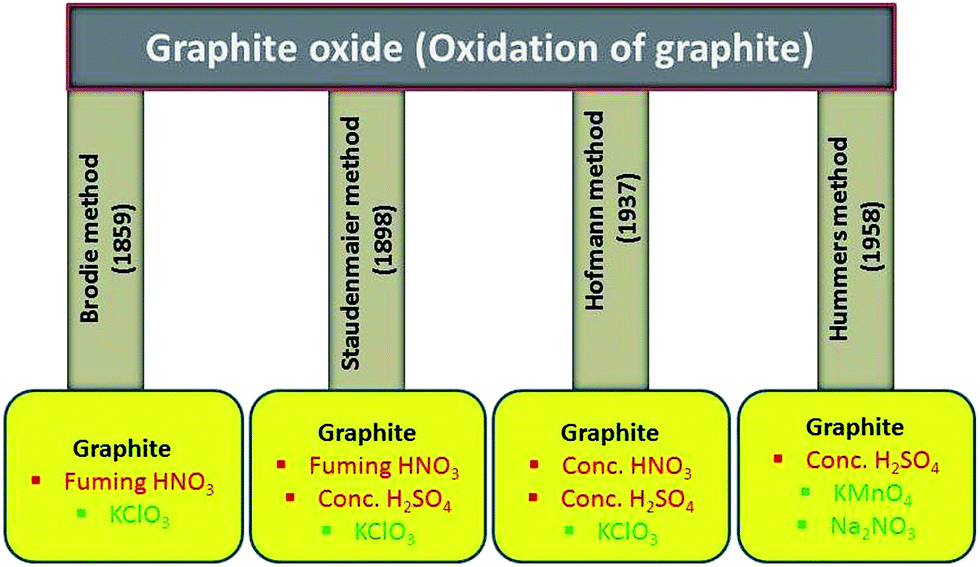 | ||
| Fig. 1 Methods for synthesis of graphite oxide using graphite, acids and oxidizing chemicals. | ||
2.5. Other methods
An improved method for the preparation of graphite oxide was reported by Marcano et al.64 in 2010 (schematic shown in Fig. 2). In this new procedure, the oxidation process was improved by excluding NaNO3, increasing the amount of KMnO4, and performing the reaction in a 9:1 mixture of H2SO4/H3PO4. This improved oxidation method significantly increases the efficiency of graphite oxidation to graphite oxide and also provides a larger amount of graphite oxide as compared to Hummers' method. This oxidation technique also prevented the formation of toxic gases (such as NO2 and N2O4). Another improved synthesis method for large-scale production of graphite oxide involves oxidation of graphite by benzoyl peroxide (strong oxidizer) at 110 °C for 10 min in an opened system.72 This technique provides a fast and efficient route to GO. Some other oxidizing agents have also been used for the preparation of GO. In this regard, Jone's reagent (H2CrO4/H2SO4) is also used for the preparation of expanded graphite, which moderately oxidizes graphite.36 The recent review by Wissler is an excellent, succinct source of further information on commonly used graphite and carbons, as well as the terminology used to describe these materials.73
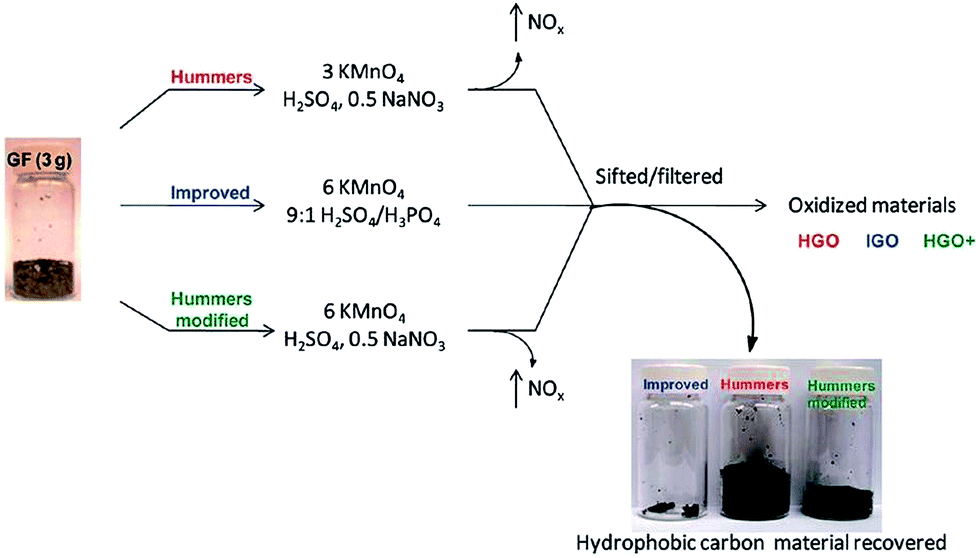 | ||
| Fig. 2 Representation of the procedures used to form GO starting with graphite flakes. Under-oxidized hydrophobic carbon material recovered during the purification of IGO, HGO, and HGO+. The increased efficiency of the IGO method is indicated by the very small amount of under-oxidized material produced.64 Reprinted (adapted) with permission from ref. 64. Copyright (2010) American Chemical Society. | ||
3. Functional groups on graphene oxide and its analysis
The structure of GO is described as graphene sheet bonded with several oxygen in the form of hydroxyl, carboxyl, epoxy and others etc. groups as shown in Fig. 3.37 Also, the structure of GO depends significantly on the purification procedures, rather than, as is commonly thought, on the type of graphite used or oxidation protocol.74 The exact identity and distribution of oxide functional groups depend strongly on the extent of oxidation. The appearance of chemical composition inside GO and the oxygen containing functional groups in GO can be identified using various techniques, including X-ray photoelectron spectroscopy (XPS),37,75–79 X-ray absorption near-edge spectroscopy (XANES),71,76,78,80,81 Fourier transform infrared spectroscopy (FTIR),78,82–84 Raman spectroscopy79,81,85 and solid-state nuclear magnetic resonance.37,84,86,87 Mkhoyan et al.41 studied the electronic and atomic structure of GO using dark field imaging of single and multilayer sheets. The results of electron energy loss spectroscopy used for measuring the structure of carbon and oxygen K-edges in a scanning transmission electron microscope indicate the high ratio of sp3 C–O bonds induces structural distortions. This suggests that the atomic structure of GO sheets should resemble a mostly amorphous 2D sheet of carbon atoms with some of them bonded to oxygen, rather than an ideal sheet of graphene with surface oxidation.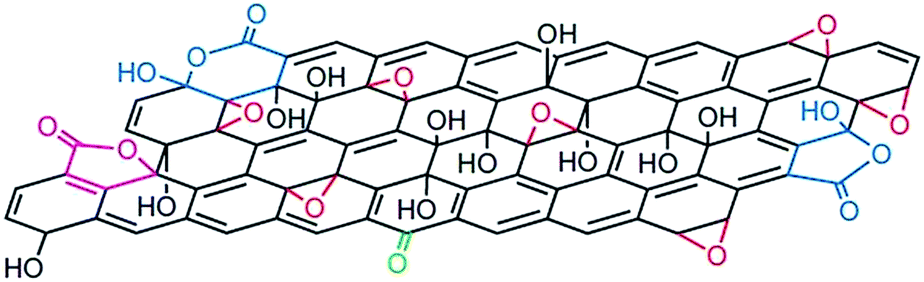 | ||
| Fig. 3 Structure of GO.37 Reprinted by permission from Macmillan Publishers Ltd: [Nature chemistry] (ref. 37), copyright (2009). | ||
Functional groups attached on GO surface can also be determined by XPS and it reveals the nature of the carbon and oxygen bonds in their various states as unoxidized carbons (sp2 carbon), C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and COOH. Several XPS investigation on GO75,76,88,89 confirms the peaks positions related with different functional groups. The deconvoluted C1s signal of GO, as seen in Fig. 4, composed of mainly five peaks (at room temperature) at different positions corresponding to sp2 carbons in aromatic rings (284.5 eV) and C atoms bonded to hydroxyl (C–OH, 285.86 eV), epoxide (C–O–C, 286.55 eV), carbonyl (〉CO, 287.5 eV), and carboxyl groups (COOH, 289.2 eV). The increase in temperature of GO shows the diminishing of the oxygen containing functional peaks and a very small fraction of CO peak remains at 1000 °C.88 The other XPS analysis,38,81 considered the deconvolution of the C1s spectra using four components, namely, sp2, C–OH, C–O–C, and COOH, while ignoring the presence of the 〉CO groups. In similar way, the deconvolution of the O1s spectra contains the main peaks around 531.08, 532.03, and 533.43 eV and these peaks are assigned to CO (oxygen doubly bonded to aromatic carbon),75,82 C–O (oxygen singly bonded to aliphatic carbon), and phenolic (oxygen singly bonded to aromatic carbon)58,90 groups, respectively. Also pristine GO shows an additional peak at a higher binding energy (534.7 eV).89
O, and COOH. Several XPS investigation on GO75,76,88,89 confirms the peaks positions related with different functional groups. The deconvoluted C1s signal of GO, as seen in Fig. 4, composed of mainly five peaks (at room temperature) at different positions corresponding to sp2 carbons in aromatic rings (284.5 eV) and C atoms bonded to hydroxyl (C–OH, 285.86 eV), epoxide (C–O–C, 286.55 eV), carbonyl (〉CO, 287.5 eV), and carboxyl groups (COOH, 289.2 eV). The increase in temperature of GO shows the diminishing of the oxygen containing functional peaks and a very small fraction of CO peak remains at 1000 °C.88 The other XPS analysis,38,81 considered the deconvolution of the C1s spectra using four components, namely, sp2, C–OH, C–O–C, and COOH, while ignoring the presence of the 〉CO groups. In similar way, the deconvolution of the O1s spectra contains the main peaks around 531.08, 532.03, and 533.43 eV and these peaks are assigned to CO (oxygen doubly bonded to aromatic carbon),75,82 C–O (oxygen singly bonded to aliphatic carbon), and phenolic (oxygen singly bonded to aromatic carbon)58,90 groups, respectively. Also pristine GO shows an additional peak at a higher binding energy (534.7 eV).89
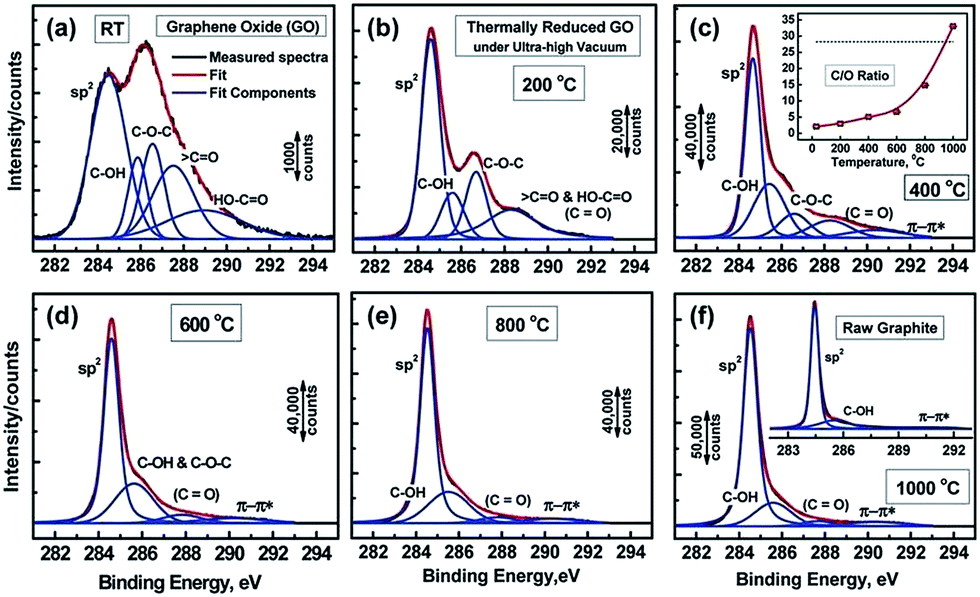 | ||
| Fig. 4 XPS spectra of GO and oxygen functionalities removal at different temperature.76 Reprinted (adapted) with permission from ref. 76. Copyright (2011) American Chemical Society. | ||
The XANES is the other powerful characterization tool for the analysis of GO materials. It provides valuable information on the degree of bond hybridization in mixed sp2/sp3 bonded carbon, specific bonding configurations of functional atoms, and degree of alignment of the graphitic crystal structures within GO.76,91
Raman spectroscopy is an experimental technique that is commonly used to characterize all sp2 carbons from three to zero dimensions, such as 3D graphite, 2D graphene, 1D carbon nanotubes, and 0D fullerenes.2 The Raman spectra of GO displays two major D band (1340 cm−1) and a broad G-band at (1580 cm−1).88 The G-band, which is characteristic of all sp2-hybridized carbon networks, originates from the first-order scattering from the doubly degenerate E2g phonon modes of graphite in the Brillouin zone center, while the prominent D peak comes from the structural imperfections created by the attachment of oxygenated groups on the carbon basal plane.92 Thus, the integrated intensity ratio of the D- and G-bands (ID/IG) indicates the oxidation degree and the size of sp2 ring clusters in a network of sp3 and sp2 bonded carbon.75,92 For example, Mattevi et al.,75 calculated the average graphitic domain size to be ∼2.5 nm in pristine GO. Lee et al.81 reported that after thermal reduction of GO, the intensity ration ID/IG significantly decreases and this indicates the considerable recovery of the conjugated graphitic framework upon removal of epoxy and hydroxyl groups. Raman spectra in Fig. 5 clearly show that the intensity of G band of rGO is high and sharp as compared to GO.
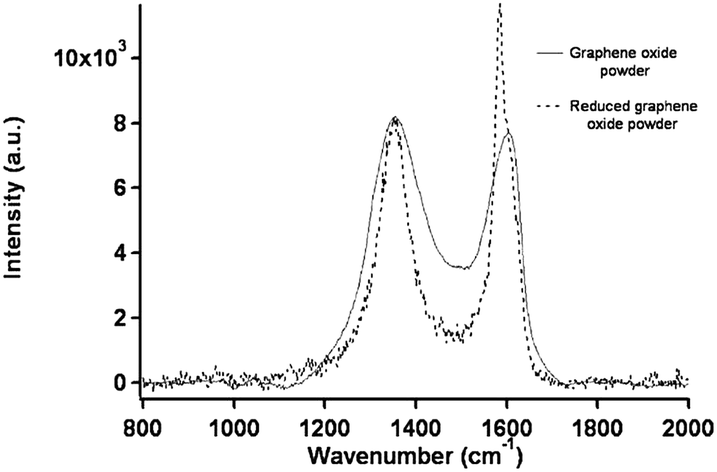 | ||
| Fig. 5 Raman spectra of the GO and rGO powder.81 Reprinted (adapted) with permission from ref. 81. Copyright (2009) American Chemical Society. | ||
FTIR spectroscopy is an important tool for characterization of functional groups attached on GO surface. Fig. 6 shows the position of absorption peaks of GO in FTIR and it indicates that different functional groups have different bond energy.78 In the case of GO, it have different peaks as hydroxyl (broad peak at 3050–3800 cm−1), carbonyl (1750–1850 cm−1), carboxyl (1650–1750 cm−1), CC (1500–1600 cm−1), and ether or epoxide (1000–1280 cm−1) groups.78,82,84
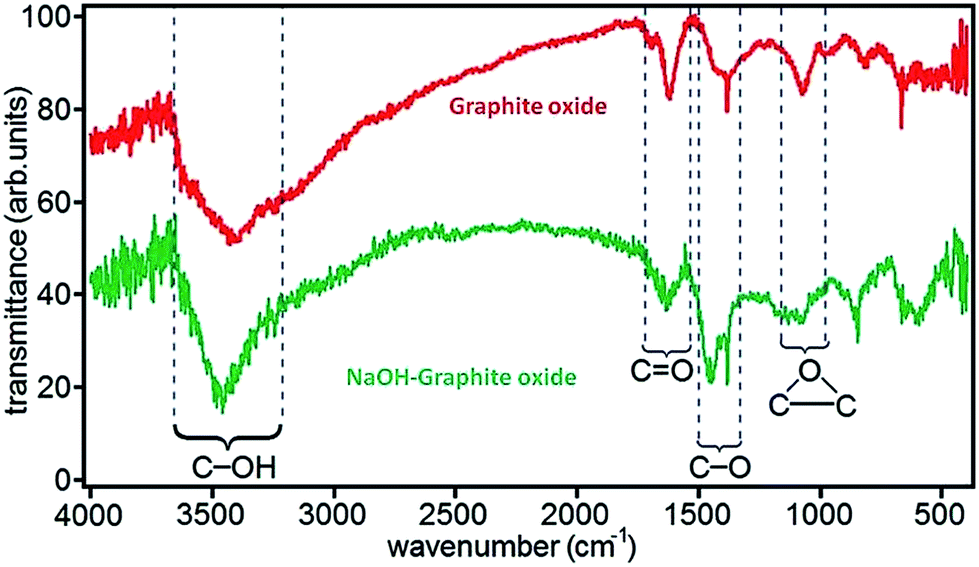 | ||
| Fig. 6 FTIR spectra of graphite oxide and NaOH–graphite oxide.78 Reprinted (adapted) with permission from ref. 78. Copyright (2010) American Chemical Society. | ||
4. Reduction of graphene oxide
4.1. Chemical reduction
Chemical reduction of graphite oxide is one of the excellent procedures to synthesized rGO and graphene in large quantities.28,93,94 It includes ultrasonication of graphite oxide in water forming a homogeneous dispersion of predominantly soluble GO in water. The GO is reduced by a suitable chemical process; the reduced GO formed resembles graphene but contains residual oxygen and other hetero atoms, as well as structural defects. During the reduction processes, most oxygen-containing functional groups of GO are eliminated and the π-electron conjugation within the aromatic system of graphite is partially restored. Finally the rGO gets precipitated from the reaction medium because of the recovered graphite domains of chemically converted graphene sheets with increased hydrophobicity and π-stacking interaction.95 The properties of rGO are nearly similar to that of graphene prepared through different chemical, thermal, photo, electrochemical or microwave reduction pathways.36 The most widely applied technique used for preparing chemically converted reduced GO is the chemical reduction of GO as shown in Fig. 7.96 | ||
| Fig. 7 Preparation of chemically converted graphene (CCG) by reduction of GO.96 Reprinted (adapted) with permission from ref. 96. Copyright © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Shin et al.97 used NaBH4 for the reduction of graphite oxide. The different molar concentrations of NaBH4 shows different order of reduction confirmed by XRD patterns and it also shows enhanced electrical properties after reduction. Fig. 8 shows the XRD pattern of graphite oxide after reduction by NaBH4 which clearly shows the shifting and broadening of peaks at different molar concentrations of NaBH4. At higher NaBH4 molar concentrations (150 mM), the peak of the large interlayer distance disappeared and shifted into a broad peak near 2θ = 23.98. This implied that functional groups were removed.
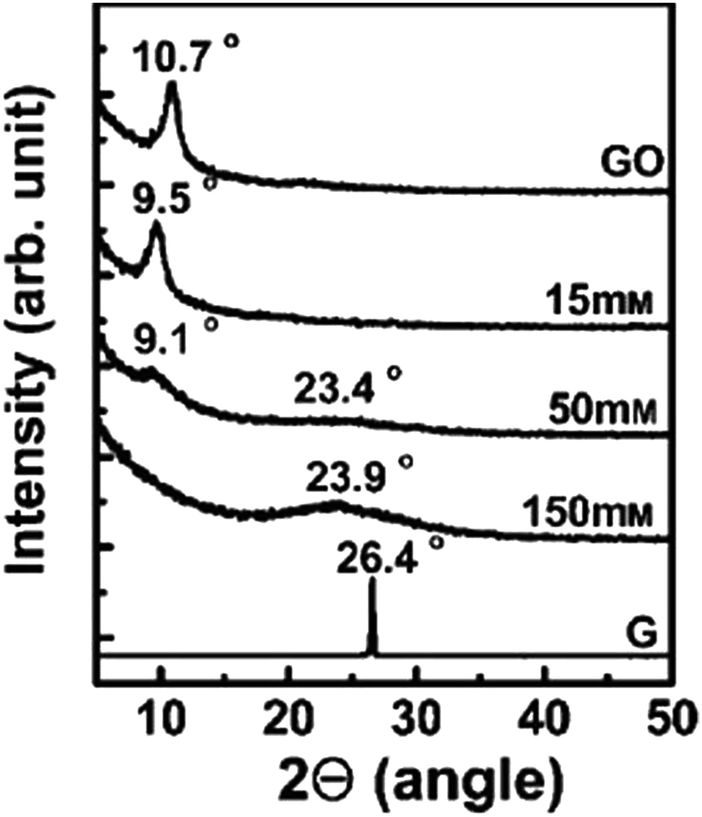 | ||
| Fig. 8 XRD patterns of GO, graphite (G), and rGO obtained using various molar concentrations of NaBH4.97 Reprinted (adapted) with permission from ref. 97. Copyright © 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Fan et al.98 reported that the exfoliated graphite oxide can undergo quick deoxygenation in strong alkali solutions like NaOH and KOH at moderate temperatures (50–90 °C) resulting in stable aqueous graphene suspensions. Fig. 9 shows that the addition of NaOH to the graphite oxide suspension led to the change in color (yellow-brown to dark black).
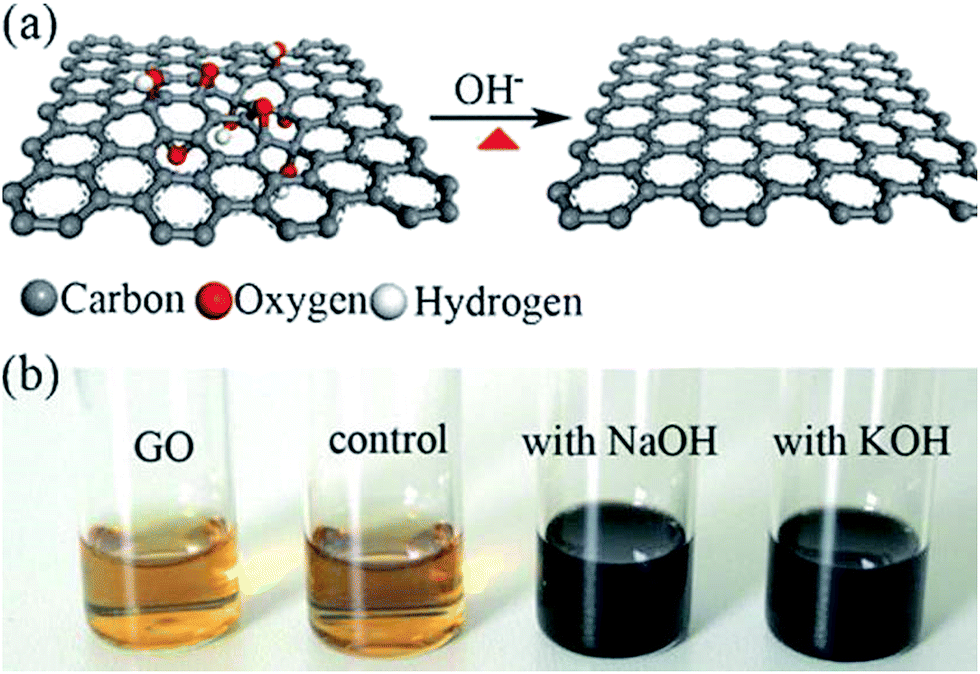 | ||
| Fig. 9 (a) Illustration for the deoxygenation of exfoliated graphite oxide under alkaline conditions and (b) images of the exfoliated-graphite oxide suspension (∼0.5 mg mL−1) before and after reaction. The control experiment in (b) carried out by heating the pristine exfoliated-graphite oxide suspension without NaOH and KOH at 90 °C for 5 h with the aid of sonication.98 Reprinted (adapted) with permission from ref. 98. Copyright © 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Various inorganic and organic reducing agents such as phenyl hydrazine,99 hydrazine hydrate,95 sodium borohydride,97,100 ascorbic acid,101,102 glucose,103 hydroxylamine,104 hydroquinone,105 pyrrole,106 amino acids,107,108 strongly alkaline solutions98 and urea109 have been explored for the chemical reduction of GO. Wang et al. reported that high-temperature alcohol vapor can also be used as an effective reducing agent of GO and the resulting chemically converted reduced graphene exhibits a highly graphitic structure and excellent electrical conductivity.105
4.2. Thermal reduction
Thermal reduction of GO or graphite oxide is an important processing step in the synthesis of many graphene-based materials and devices. The GO reduced by heating is known as thermal reduction by annealing. Annealing atmosphere is also important for the reduction of GO. Annealing reduction is usually carried out in vacuum,110 or in inert111 or reducing atmosphere.111–113 Thermal reduction of GO comprised of the thermal-energy-induced multistep removal of intercalated H2O molecules and oxide groups of carboxyl, hydroxyl, and epoxy. It should be noted that in chemical reduction, individual GO sheets in the solution phase are chemically reduced by the strong chemical base.114 The rapid heating of graphite oxide at high temperature, exfoliates in the form of porous carbon materials and get converted into graphene with fewer amounts of oxygen functionalities. The exfoliation occurs by the sudden expansion of CO or CO2 gases evolved from the spaces between graphene sheets during rapid heating of the graphite oxide. The rapid heating makes the oxygen containing functional groups attached on carbon plane to decompose into gases that create huge pressure between the stacked carbon layers. Based on state equation, a pressure of 40 MPa is generated at 300 °C, while 130 MPa is generated at 1000 °C.115 The evaluation of the Hamaker constant predicts that a pressure of only 2.5 MPa is enough to separate two stacked GO platelets.115 Rapid heat treatment at elevated temperature, not only exfoliate graphite oxide but also reduce the functionalities by decomposing oxygen containing groups. The notable effect found during the thermal exfoliation is the structural defect and damage of graphene sheets caused by the release of carbon dioxide.90 These structural defects and damage inevitably affect the electronic properties by decreasing the ballistic transport path length and introducing scattering centers.The heating temperature severely affects the removal of oxygen from GO surfaces and consequently effect the reduction of GO.90,110,111 Li et al.112 monitored the variation of chemical structure with annealing temperature, and concludes that the high temperature is needed to achieve the better reduction of GO. Schniepp et al.90 reported the C/O ratio less than 7 for heating temperature less than 500 °C, and its ratio increases to higher than 13 as the heating temperature reaches to 750 °C. The reduction of GO usually enhances the electrical conductivity due to the removal of oxygen containing groups. Wang et al.111 found the conductivity of GO thin films to increases with annealing temperature as shown in Fig. 10. Electrical conductivity of the reduced GO film obtained at 550, 700, 900 and 1100 °C were 49, 93, 383 and 550 S cm−1, respectively. Wu et al.113 reported the hydrogen arc-discharge exfoliation technique for exfoliation of graphite oxide to prepare graphene. Since the arc-discharge could provide temperatures above 2000 °C in a short time and the electrical conductivity of graphene sheets was found to be 2000 S cm−1.
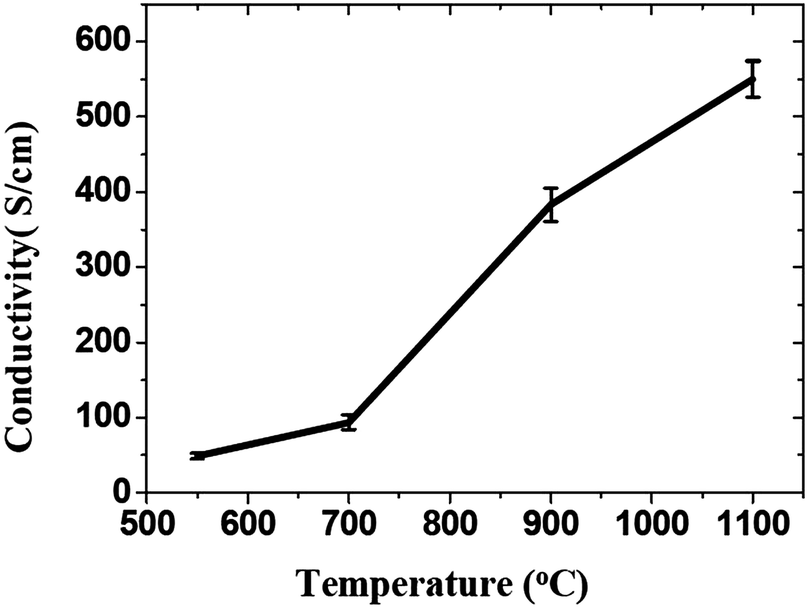 | ||
| Fig. 10 Increase of the average conductivity of graphene films with temperature.111 Reprinted (adapted) with permission from ref. 111. Copyright (2008) American Chemical Society. | ||
4.3. Microwave and photo reduction
Microwave heating is widely used for the exfoliation and reduction of graphitic precursor materials. With microwave treatments, the preparation of graphene can be performed by solid-state reduction of dry graphite oxide, the reduction of graphite oxide in suspension, or by heating graphite intercalated compounds (GICs). Furthermore, the simultaneous reduction and doping of GO and the preparation of hybrid materials is also possible. The GO synthesis and reduction using unconventional heating like microwave-irradiation (MWI)116–118 and photo-irradiation119,120 is a fast and energy-efficient method. MWI induced heating has been used as a rapid way to synthesize graphene sheets.Microwave assisted heating can provide significant enhancement in the transfer of energy directly to the reactants which causes instantaneous rise in internal temperature.118,121 The rise in instantaneous internal temperature can shorten the reaction time significantly, and improves the reaction efficiency. Also, owing to the difference in the solvent and reactant dielectric constants, selective dielectric heating provides significant enhancement in the direct energy transfer to the reactants, which causes an instantaneous internal temperature rise and thereby reduction of GO.118 Dry GO absorbs MWI strongly with a sudden increase in surface temperature up to ∼400 °C, of the GO, within just 2 s, leading to an ultrafast reduction of GO to rGO.103 Several research groups have used MWI for the synthesis as well as reduction of GO.117,122–126 Zhu et al.117 reported a simple and versatile method to simultaneously achieve the exfoliation and reduction of graphite oxide within 1 min by treating graphite oxide powders in a commercial microwave oven. Yan et al.127 described a new approach for the reduction of GO using glucose in an alkaline environment by microwave heating. Chen et al.128 reduces GO in a mixed solution of N,N-dimethylacetamide and water (DMAc/H2O). The mixed solution works both as a solvent for the production of graphene and as a medium to control the temperature of the reactive system up to 165 °C. Here the reduction of GO can be accomplished in time scale of minutes, and the degree of reduction depends on the duration of microwave treatment and given power (800 W). Fig. 11 shows the optical image of GO suspension with concentration of 0.56 mg mL−1 in water or DMAc/water mixture before and after microwave heating for different times. At first, an aqueous suspension of GO is yellow-brown in color and after microwave irradiation for different time duration its color starts to change and finally changes to black. The GO suspension exhibited long-term stability at room temperature without precipitation.
 | ||
| Fig. 11 Photographs of GO suspension with concentration of 0.56 mg mL−1 (a–f) under different conditions. (a) GO aqueous suspension after microwave treatment for 1 min, (b) GO suspension in DMAc/H2O (6:1), (c–f) GO suspension in DMAc/H2O (6:1) after microwave treatment for 1, 2, 3, and 10 min (2 min × 5 times), respectively, (g) re-dispersed of the dried graphene into pure DMAc with concentration of 1.0 mg mL−1.128 Reprinted (adapted) with permission from ref. 128. Copyright © 2010 Elsevier B. V. All rights reserved. | ||
Photo reduction like photo-thermal and photo-chemical reduction of GO is a additive free, facile, clean, and versatile approach to reduce GO. High-quality rGO has been prepared by irradiating GO with sunlight, ultraviolet light, and KrF excimer laser.129 Ding et al.130 reduced GO using UV irradiation to obtain single to few-layered graphene sheets without the use of any photocatalyst. Cote et al.120 prepared rGO by photothermal reduction of GO using xenon flash at ambient conditions and patterned GO or GO/polymer films using photomask. Nanosecond laser pulses of KrF eximer laser of 335 and 532 nm effectively reduces dispersed GO to thermally and chemically stable graphene.131 Photochemical reduction of GO to graphene has also been exploited for patterning. The photo-reduction and patterned film fabrication was carried out with femto-second laser irradiation.119 The focused laser beam (laser pulse of 790 nm central wavelength, 120 fs pulse width, 80 MHz repetition rate, focused by a ×100 objective lens) was used to heat the localized GO film with a line width in the range of 1.5 μm. As a result, the laser reduction can produce rGO films with a much higher conductivity with laser incident power by laser writing as shown in Fig. 12.
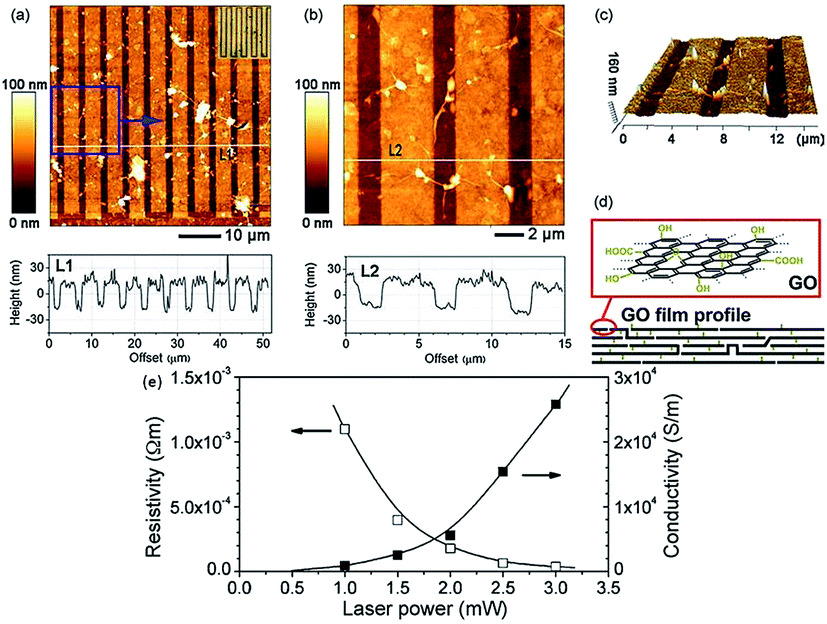 | ||
| Fig. 12 (a) AFM image of comb-like microcircuit and height profile along the white line (L1), inset is the optical micrograph of the microcircuit; (b) magnified image of the local pattern (blue square of a) and height profile along the white line (L2); (c) 3D image of (b); (d) illustration for the profile of GO film before and after femto second laser reduction and (e) dependence of resistivity and conductivity of reduced and pattern GO micro-belts on laser power.119 Reprinted (adapted) with permission from ref. 119. Copyright © 2005 Elsevier B. V. All rights reserved. | ||
Supported GO films on different substrates were also irradiated by excimer laser for its reduction. GO film deposited on Si substrates were subjected to excimer laser radiation (Lambda Physik KrF excimer laser, 248 nm wavelength, 30 ns lifetime, 300 mJ laser energy, 5 Hz repetition rate, 200 shots), for reduction and patterning.132,133 The high energy direct electron-beam is also capable for the reduction of GO.134 The GO can be reversibly reduced and oxidized using electrical stimulus. Controlled reduction and oxidation in two-terminal devices containing multilayer GO films was demonstrated by Ekiz et al.135 and Yao et al.136
4.4. Photo catalyst reduction
The review article published by Pei et al. gives a detailed overview and description about the reduction of GO by photocatalysis method.94 The attached functional groups on GO surfaces can be removed by the help of photocatalyst like TiO2 which turns it into reduced GO. Kamat research group137 reported the reduction of GO in a colloidal state with the help of TiO2 nanoparticles under ultraviolet irradiation. The GO suspended in ethanol undergoes reduction as it accepts electrons from UV-irradiated TiO2 nanoparticles suspensions. After reduction through TiO2 photo catalyst, the color of the GO suspension changes from brown to black as shown in Fig. 13. Also the photocatalytic properties of semiconducting TiO2 particles have been thoroughly investigated.138 In the presence of ethanol the holes are scavenged to produce ethoxy radicals, thus leaving the electrons to accumulate within the TiO2 particles. The accumulated electrons interact with GO sheets to reduce functional groups and before reduction, the carboxyl groups in GO surface can interact with the hydroxyl groups on the TiO2 surface by charge transfer mechanism, producing a hybrid connection between TiO2 and the GO, and this structure can be retained after reduction. The rGO sheets can work as a current collector to facilitate the separation of electron/hole pairs in some photovoltaic devices like photocatalysis device139 and in dye-sensitized solar cell.140,141 The similar reduction mechanism has also been found in other carbon materials such as carbon nanotubes and fullerene.142,143 Beyond TiO2, different materials with photo catalytic activity, like BiVO4 (ref. 128) and ZnO144 have also been reported to achieve the reduction of GO.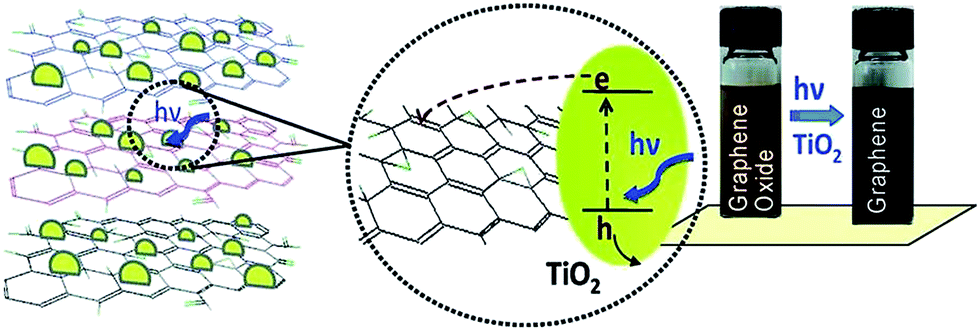 | ||
| Fig. 13 Photo catalyst electron acceptance by GO and change in color from brown to black after GO reduction.137 Reprinted (adapted) with permission from ref. 137. Copyright (2008) American Chemical Society. | ||
4.5. Solvothermal/hydrothermal reduction
The reduction of GO through solvothermal or hydrothermal route generally occurs at low temperature and high pressure is also an important series in the field of graphene synthesis and reduction.145–147 Solvothermal processes can be defined as chemical reactions or transformations in a solvent under supercritical conditions or near by pressure–temperature domain resulting from heating.148 Inside the sealed container, the reduction occurs through processes involving surface chemistry, the reactivity being increased under high pressure conditions and at moderate temperatures. The hydrothermal method has been applied for the transformation of carbohydrate molecules to form homogeneous carbon nano-spheres149 and nanotubes.150 Zhou et al.147 reported that supercritical water plays the role of reducing agent in hydrothermal conditions and offers green chemistry alternative to organic solvents. This result shows that the supercritical water removes the functional groups attached on GO and also recovers the aromatic structures of carbon lattice. Fig. 14 shows that after reduction the solution color changes from brown to dark black and UV visible absorption shows the decrease in band gap after reduction of GO.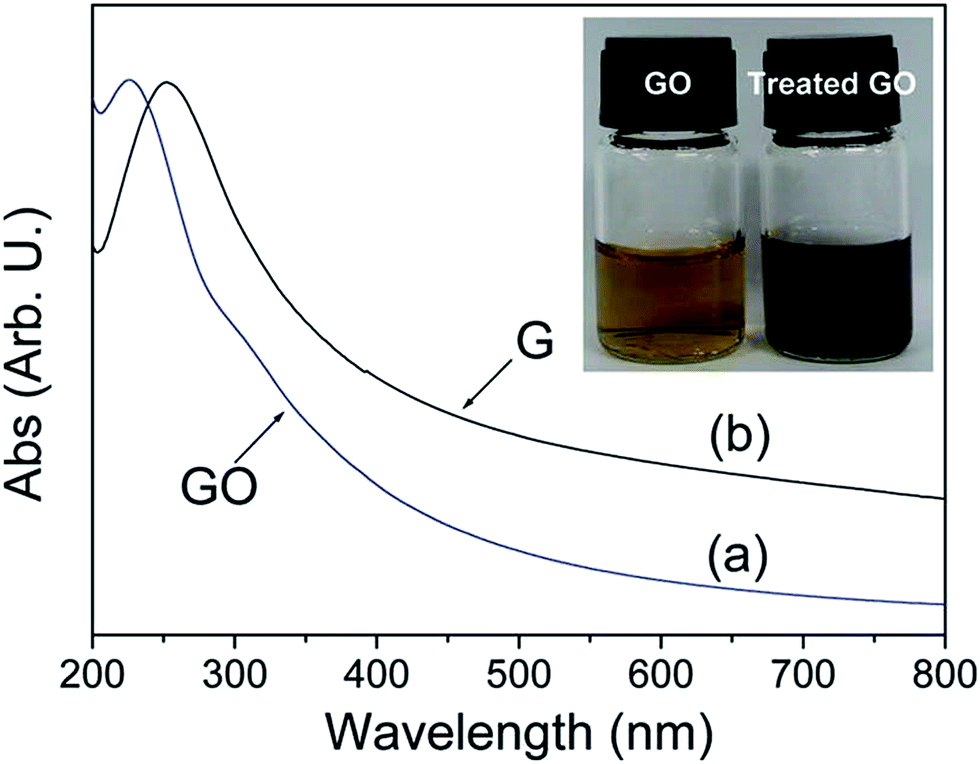 | ||
| Fig. 14 UV visible absorption spectra of the GO (a) before and (b) after hydrothermal treatment at 180 °C for 6 h. Inset shows the color change of 0.5 mg mL−1 GO solution before and after hydrothermal treatment.147 Reprinted (adapted) with permission from ref. 147. Copyright (2009) American Chemical Society. | ||
Wang et al.146 employed N,N-dimethylformamide as solvent for the reduction of GO using solvothermal method. After solvothermal treatment (180 °C for 12 h) the C/O ratio of rGO is higher than that produced by hydrazine reduction at normal pressure. It reveals that more oxygen content was removed after the solvothermal treatment. Dubin et al.145 proposed a modified solvothermal reduction method using N-methyl-2-purrolidinone (NMP) as solvent. This treatment was not performed in a sealed container and the heating temperature was lower than the boiling point of NMP. As a result, there is no additional pressure present in the reduction process. The C/O ratio of the solvothermal rGO is very low (5.15) and this reduction method can only achieve a moderate reduction of GO. The electrical conductivity of the rGO films achieved by this solvothermal reduction and subsequent vacuum filtration is 3.74 × 102 S cm−1, which is one order of magnitude smaller than that produced by hydrazine reduction (8.28 × 103 S cm−1).
Various research groups have shown the reduction of GO using different chemical agents with different solvent, at different temperatures and time as summarised in Table 1.
| Reducing chemical agents/method | Temp. (°C) | Time (h) | Solvent | O content (at%)/C/O atomic ratio* | Ref. |
|---|---|---|---|---|---|
| N2H4 + NH3 solution | 95 | 1 | H2O | — | 151 |
| N2H4/DMF | 180 | 12 | DMF | 1.5 | 146 |
| Ascorbic acid | RT | 48 | H2O | — | 101 |
| Hexamethylenetetramine | RT | 12 | H2O | — | 152 |
| S-Containing compound | 95 | 3 | H2O/DMAc–H2O | 6.48* | 153 |
| Glucose + ammonia solution | 95 (RT) | 1 (48) | H2O | — | 154 |
| Hydrazine hydrate | 100 | 24 | H2O | 10.3* | 95 |
| Fe powder | RT | 6 | H2O | 7.9* | 155 |
| Zn + H2SO4 | RT | 2 | H2O | 4.5 | 156 |
| Hydroquinone | Refluxed | 20 | H2O | — | 105 |
| NaBH4 | 125 | 3 | H2O | — | 72 |
| Tellurium nanowire | RT | 36 | H2O | — | 157 |
| Ascorbic acid + amino acid | 80 | 24 | H2O | — | 107 |
| Solvothermal reduction | 205 | 24 | NMP | 13.8 | 145 |
| Solvothermal reduction | 120–200 | 4–48 | H2O, ethanol, 1-butanol, ethelene glycol | 6.6–16.6 | 158 |
| Thermal reduction | 150 | 8 | DMAc + H2O | 4.7* | 159 |
5. Frontier applications of graphene oxide
5.1. Lithium ion battery application
Anode based on hydrogen-enriched reduced GO showed an enhanced electrochemical performance as reported by Yoon et al.168 A sandwiched nano-architecture of rolled-up Si/reduced GO bilayer nano-membranes was utilized as anodes for lithium-ion batteries, which demonstrated long cycling life of 2000 cycles at 3 A g−1 with a capacity degradation of only 3.3% per 100 cycles.169 Zeng et al.170 presented a facile approach to fabricate flexible spinel-type oxide/reduced GO composite aerogels as binder-free anodes. Gao et al.171 reported a multi-layered structural silicon-reduced GO electrode which showed a stable charge–discharge performance with high rate, a reversible capacity of 2787 mA h g−1 at a charging rate of 100 mA g−1, and a stable capacity over 1000 mA h g−1 retained at 1 A g−1 after 50 cycles. The cycling performance and columbic efficiency of the Si/rGO anode are shown in Fig. 15a. Fig. 15b and c shows galvanostatic cycling and the rate capability respectively.
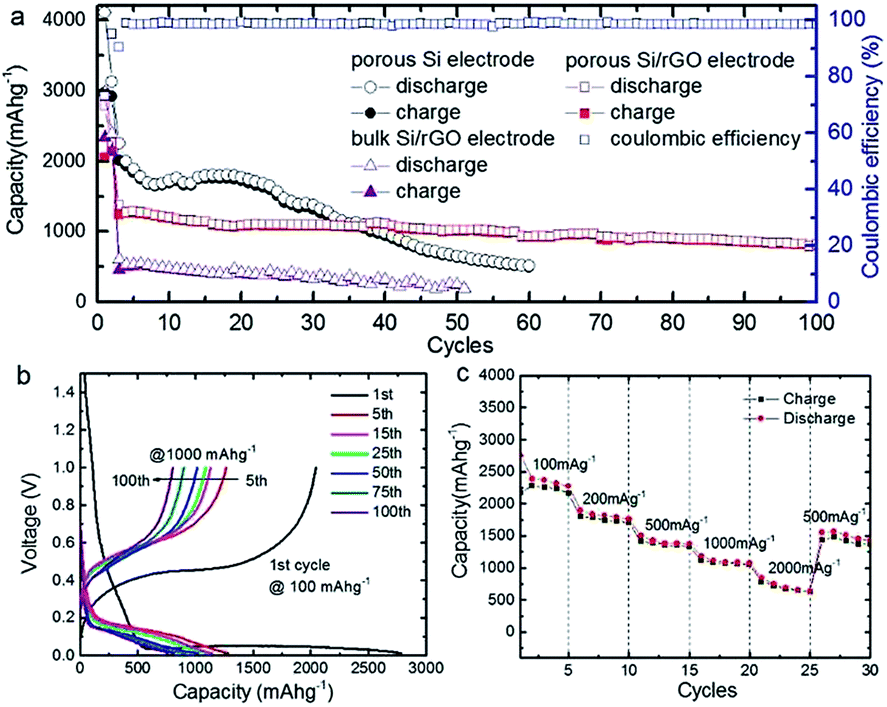 | ||
| Fig. 15 (a) Cycle performance at a constant current rate of 1000 mA g−1 after initial activation processes at 100 mA g−1 for two cycles; (b) galvanostatic discharge–charge profiles; and (c) rate performance of Si/graphene composite electrode.171 Reprinted (adapted) with permission from ref. 171. Copyright (2015) American Chemical Society. | ||
Besides various rGO based composites like (i) crumpled rGO/MoS2 nanoflowers,172 (ii) amorphous GeOx-coated rGO balls,173 (iii) copper sulfide nanowires/rGO,174 (iv) Si/Ti2O3/rGO,175 (v) MnS hollow microspheres-rGO,176 (vi) copper silicate hydrate hollow spheres-rGO177 (vii) fluorine-doped tin oxide nanocrystal/rGO,178 (viii) hollow nanobarrels of α-Fe2O3 on rGO179 and (ix) hierarchical rGO-Co2V2O7 nanosheets180 etc. shows an improved performance and stability as anode material for the lithium-ion batteries.
A generic method of coating GO on particles by engineering the ionic strength of solutions was developed by Rong et al.185 Uniform coating of GO on various particles with a wide range of sizes, geometries, and compositions were achieved. GO on sulfur particles to form sulfur/GO core–shell particles as Li–S battery cathode materials was investigated, and found that the sulfur/GO composite material exhibits significant improvements in electrochemical performance over sulfur particles without coating. The optical images and charge discharge curves of the sulfur/GO are shown in the Fig. 16.
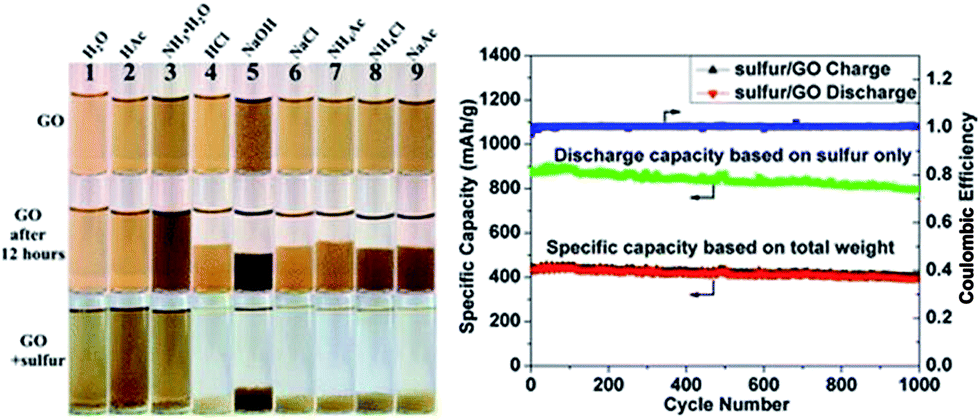 | ||
| Fig. 16 Digital images of GO dispersed in different solutions at the beginning, after 12 h and after adding sulfur particles to GO dispersion. Galvanic charge–discharge performance and coulombic efficiency of sulfur/GO at 1 A g−1 for 1000 cycles. Discharge specific capacity calculated based on weight of sulfur only (green dotted line) and total weight of sulfur/GO are plotted.185 Reprinted (adapted) with permission from ref. 185. Copyright (2014) American Chemical Society. | ||
Since lithium sulfide (Li2S) is a promising cathode material for Li–S batteries with high capacity (theoretically 1166 mA h g−1) and can be paired with non lithium–metal anodes to avoid potential safety issues. However, the cycle life of coarse Li2S particles suffers from poor electronic conductivity and polysulfide shuttling. Wang et al.186 developed a flexible slurryless nano-Li2S/reduced GO cathode paper which was directly used as a free-standing and binder-free cathode without metal substrate. It showed an excellent rate capability and cycle life. Hwa et al.187 also reported the lithium sulfide/GO nanospheres with conformal carbon coating as a high-rate, long-life cathode for Li/S cells. Huang et al.188 proposed a unique lithium sulfur battery configuration with an ultrathin GO membrane for high stability.
700 mA h g−1, a low charge/discharge overpotential (about 0.355 V), and a long cycle life up to 200 cycles (under the curtaining capacity of 1000 mA h g−1).91 Nanocrystalline Co3O4, grown on reduced graphene oxide (Co3O4/RGO) was employed as part of a carbon-based oxygen electrode membrane which results in significant reduction of overpotentials for OER (up to 350 mV), and improved cycling performance in the Li–O2 cell.190 Li et al.191–193 employed pure, nitrogen and sulphur doped graphene nano sheets derived from the thermal reduction of GO in lithium–oxygen batteries and found that they show excellent electrocatalytic activity for oxygen reduction.5.2. Solar cell applications
Organic photovoltaic (OPV) materials have recently garnered significant attention as enablers of high power conversion efficiency (PCE), low-cost, mechanically flexible solar cells. Nevertheless, further understanding-based materials developments will be required to achieve full commercial viability. In particular, the performance and durability of many current generation OPVs are limited by poorly understood interfacial phenomena.The utilization of GO thin films as the hole transport and electron blocking layer in organic photovoltaics was demonstrated by Li et al.194 The incorporation of GO deposited from neutral solutions between the photoactive poly(3-hexylthiophene) (P3HT):phenyl-C61-butyric acid methyl ester (PCBM) layer and the transparent and conducting indium tin oxide led to a decrease in recombination of electrons–holes and leakage currents. This resulted in a dramatic increase in the OPV efficiencies to values that are comparable to devices fabricated with PEDOT:PSS as the hole transport layer. Liu et al.195 utilized sulfated GO as a hole-extraction layer for high-performance polymer solar cells. Radich et al.196 reported Cu2S reduced GO composite for high-efficiency quantum dot solar cells.
Murray et al.197 applied electronically tuned GO as an effective interfacial layer (IFL) for templating the optimum donor polymer π-stacking orientation for high efficiency OPVs. In addition to functioning as a solution-processable PEDOT:PSS alternative, GO significantly enhances the durability of fully fabricated devices by increasing the active layer-IFL interfacial stability under thermal and environmental stress.
Kavan et al.198 used optically transparent cathode for Co(III/II) mediated dye-sensitized solar cells (DSSC) based on GO. Various other counter electrodes like (i) rGO–TaON composite for Co(bpy)33+/2 mediated DSSC,199 (ii) multiwalled carbon nanotube@rGO nanoribbon,200 (iii) TiO2 photo-anodes with nitrogen–rGO,201 (iv) rapid atmospheric pressure plasma jet processed rGO,202 (v) nanocomposite of tin sulfide nanoparticles with rGO,203 and (vi) GO assisted anatase TiO2 nanosheets204 were utilized to improve the overall performance and efficiencies of the solar cells. Han et al.205 reported rGO/mesoporous–TiO2 nanocomposite based meso-structured perovskite solar cells that showed an improved electron transport property owing to the reduced interfacial resistance. This effect significantly increased the short circuit current density and open circuit voltage. The rGO/mesoporous–TiO2 nanocomposite film with an optimal rGO content of 0.4 vol% shows 18% higher photon conversion efficiency compared with the TiO2 nanoparticles based perovskite solar cells.
5.3. Supercapacitor applications
An alternative and effective route to prepare conducting polyaniline-grafted rGO composite with highly enhanced properties was reported by Kumar and co-workers.206 The composite showed more pronounced electrochemical performances than the pristine GO as supercapacitor electrodes, exhibiting a capacitance value of 250 F g−1. Xu et al.207 prepared GO (GO)/polyaniline nanocomposite by in situ polymerization with the assistance of supercritical carbon dioxide (SC CO2) which exhibited high specific capacitance (425 F g−1) at a current density of 0.2 A g−1. A hierarchical high-performance electrode with nanoacanthine-style polyaniline deposited onto a carbon nanofiber/GO template was successfully prepared by Xu et al.208 via in situ polymerization process. The hierarchical free-standing electrodes showed a significant specific capacitance of 450.2 F g−1 at the voltage scan rate of 10 mV s−1.Zhang et al.209 developed a highly conductive, free-standing, and flexible porous carbon film by simple KOH activation of reduced G-O films. The supercapacitors constructed with these films as electrodes yielded outstanding power and frequency performance, simultaneously maintaining excellent energy densities. Li et al.210 prepared hierarchical flowerlike NiO/reduced GO composites which showed a maximum specific capacitance ∼428 F g−1 at a discharge current density of 0.38 A g−1 in a 6.0 M KOH electrolyte.
Hsieh et al.211 reported high specific capacitance, superior rate capability, and excellent cyclic stability of reduced GO nanosheets decorated with SnO2 nanocrystals as electrode materials for electrochemical capacitors. Xing et al.212 reported a facile photochemical reduction method for producing the photoreduced graphene hydrogels with 3D porous structures from GO suspension. The supercapacitor based on this structure performed a high specific capacitance of 254 F g−1 at 1 A g−1 in KOH electrolyte. Sahu et al.213 synthesized high performance lacey rGO nanoribbons (LrGONR). Holes created during the LrGONR synthesis not only enhanced the electrolytic accessibility but destacked all the graphene layers through protrusion at edge planes and corrugation in individual graphene. The supercapacitor exhibited exceptionally high energy/power density, typically 15.06 W h kg−1/807 W kg−1 in aqueous at 1.7 A g−1, 90 W h kg−1/2046.8 W kg−1 in non aqueous at 1.8 A g−1, and 181.5 W h kg−1/2316.8 W kg−1 in ionic electrolyte at ∼1.6 A g−1.
A ternary nanocomposite system, composed of poly(vinylidene fluoride) (PVDF), NH2-treated graphene nanodots (GND), and rGO, was synthesized by Sunghun Cho et al.214 The resulting PVDF/NH2 treated GND/rGO nanocomposite exhibited higher dielectric constant (ε′ ≈ 60.6) and larger energy density (Ue ≈ 14.1 J cm−3) compared with the pristine PVDF (ε′ ≈ 11.6 and Ue ≈ 1.8 J cm−3).
Kumar et al.55,190–193,215 synthesized different graphene–metal oxide based hybrid materials as electrodes for supercapacitor applications. The reported work shows that the electrochemical performance of a interesting three-dimensional structures comprised of zero-dimensional cobalt oxide nanobeads, one-dimensional carbon nanotubes and two-dimensional graphene, stacked hierarchically.215 The electrode based on this hierarchical structure showed an electrochemical performance with a maximum specific capacitance of 600 F g−1 at the charge/discharge current density of 0.7 A g−1 in KOH electrolyte. An ultrathin supercapacitor electrode was fabricated by solution-processable assembly of rGO nanosheets with a photo-cross-linkable polymer by Jo et al.216 UV treatment triggered cross-linking of DR with PEDOT:PSS, causing pure electrostatic interactions to convert to covalent bonds. This film exhibited a large volumetric capacitance value of 354 F cm−3.
Three dimensional rGO/Ni foam composites were prepared by Yang et al.217 by reduction of GO by Ni foam directly in its aqueous suspension at pH 2 at room temperature. When the reduction time increased from 3 to 15 days, the areal capacitance of the composite increases from 26.0 to 136.8 mF cm−2 at 0.5 mA cm−2. The 5 h rGO/Ni foam composite shows an areal capacitance of 206.7 mF cm−2 at 0.5 mA cm−2 and good rate performance and cycling stability with areal capacitance retention of 97.4% after 10000 cycles at 3 mA cm−2. Further extending the reduction time to 9 h at 70 °C, the composite shows a high areal capacitance of 323 mF cm−2 at 0.5 mA cm−2. Fig. 17a shows the digital image of Ni foams before and after 7 day immersion in the GO aqueous suspension with different pH values. The microstructures of pristine Ni foam and the rGO/Ni foam composite prepared at pH-2 for 7 days are shown in Fig. 17b–d.
 | ||
| Fig. 17 (a) Digital image of pristine Ni foam and the Ni foams after immersion in GO suspensions with different pH values for 7 days of reduction at room temperature. SEM images of (b) pristine Ni foam, (c, d) freeze-dried 7 day rGO/Ni foam composite, and (e) freeze-dried 3 day rGO/Ni foam composite.217 Reprinted (adapted) with permission from ref. 217. Copyright (2016) American Chemical Society. | ||
5.4. Bio applications
Being the newest member of the carbon materials family, graphene possesses many unique physical properties resulting is a wide range of applications. Recently, it was discovered that GO can effectively adsorb DNA, and at the same time, it can completely quench adsorbed fluorophores. These properties make it possible to prepare DNA-based graphene materials for various bio applications in detection and cure of the diseases.Wu et al.218 described fluorescent-labeled oligonucleo-tides on GO surface. The binding between DNA and GO is strongly affected by both electrostatic repulsion and hydrophobic interactions. Cations, pH, and organic solvent can all be used to modulate DNA binding. Shorter DNA binds to the surface with faster kinetics and higher adsorption efficiency. Desorption can occur by adding the c-DNA to form ds-DNA, adding the same DNA to exchange, and increasing temperature. The temperature-induced desorption, however, was quite ineffective, suggesting a high binding affinity. Overall, the DNA binding to GO is very stable and reversible. These basic understandings of the surface interaction between DNA and GO are described in the Fig. 18. The mechanism of DNA adsorption and desorption on GO was explained by Park et al.219
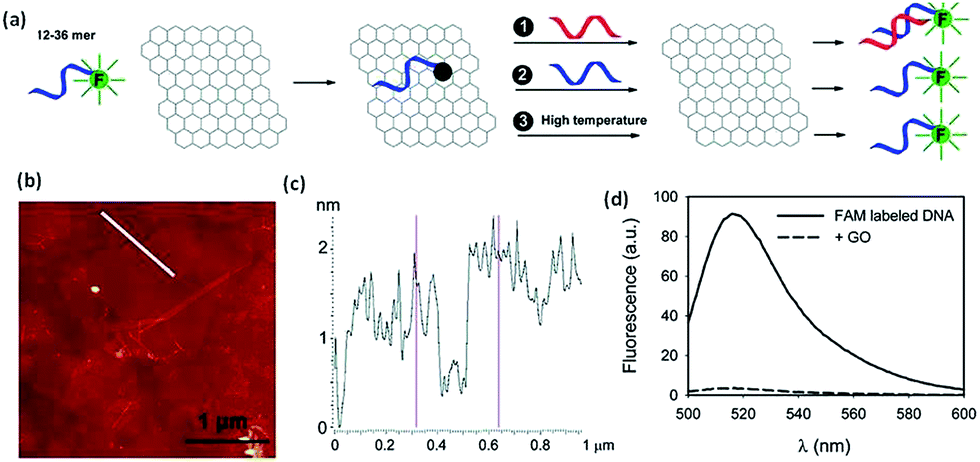 | ||
| Fig. 18 (a) Schematic presentation of FAM-labeled DNA adsorption and desorption on GO. Fluorescence is quenched upon adsorption. Desorption can be achieved by adding the c-DNA (reaction 1), DNA exchange with the same DNA (reaction 2), or increasing temperature (reaction 3). (b) An AFM image showing GO sheets deposited on a silicon wafer. (c) The height profile of the line in (b) shows the sheets to be ∼1.5 nm in thickness. (d) Fluorescence spectra of 100 nM 18-mer DNA in 25 mM HEPES, 100 mM NaCl, and 5 mM MgCl2 in the presence and absence of 50 μg mL−1 of GO.218 Reprinted (adapted) with permission from ref. 218. Copyright (2011) American Chemical Society. | ||
Zhang et al.220 demonstrated that uniform ultrasmall GO nanosheets has very low cytotoxicity and high cellular uptake. Therefore, the as-prepared uniform ultrasmall GO nanosheets could be explored as the ideal nanocarriers for drug delivery and intracellular fluorescent nanoprobe. Liu et al.221 reported transferrin modified GO for glioma-targeted drug delivery and was evaluated in vitro and in vivo also. Zhang et al.222 developed a GO-DTPA-Gd/DOX-based theranostic nanoplatform with dual modality T1 MRI/fluorescence imaging and drug delivery functionalities. Weaver et al.223 reported electrically controlled drug delivery nanocomposite composed of GO deposited inside a conducting polymer scaffold.
Various graphene based composites like (i) patterned substrates of nano-GO224 (ii) GO/hydrogel-based angiogenic225 (iii) hyaluronic acid-decorated GO nanohybrids226 (iv) functionalized GO nanoparticles,227 and (v) fluorescent GO via polymer grafting227 etc. were utilized for efficient drug delivery. GO could be fabricated by modifying its functional groups to impart specific functional or structural attributes. This study demonstrated the development of a GO-based efficient gene delivery carrier through installation of polyethylenimine, a cationic polymer, which has been widely used as a non viral gene delivery vector. Kim et al.228 demonstrated the successful fabrication of GO through covalent linkage with LMW BPEI, which acted as a cationic gene carrier. The BPEI-GO possessed high gene delivery efficiency and exhibited high cell viability. Furthermore, the PL properties of GO were enhanced through conjugation of BPEI to GO, which advocated the immense potential of BPEI-GO as a fluorescence reagent. Gold encapsulated nanoparticles or nanorods in GO shells showed a great response as a novel gene vector.229
Zhang et al.230 grew porous platinum nanoparticles on GO which was proven to function as peroxidase mimetics that can catalyze the reaction of peroxidase substrate in the presence of hydrogen peroxide. On the basis of the peroxidase-like activity, this composite was utilized as a signal transducer to develop a colorimetric assay for the direct detection of cancer cells as described in the Fig. 19.
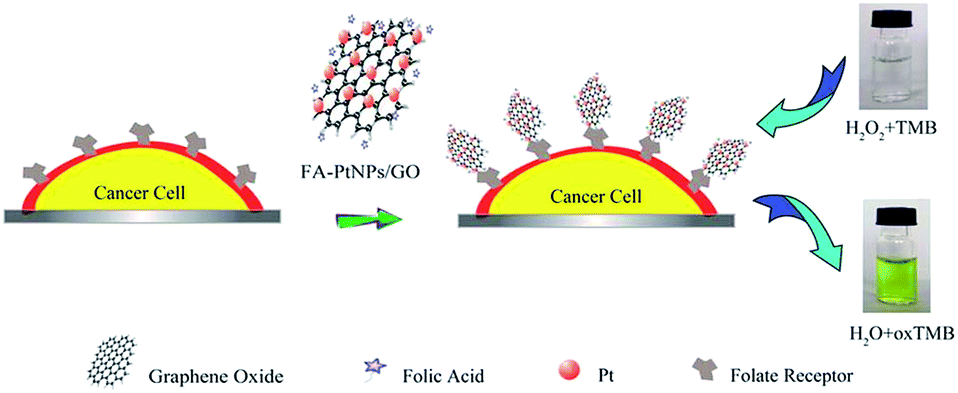 | ||
| Fig. 19 Schematic representation of colorimetric direction of cancer cells by using folic acid functionalized PtNPs/GO.230 Reprinted (adapted) with permission from ref. 230. Copyright (2011) American Chemical Society. | ||
Fluorogenic resveratrol-confined GO were utilized for the detection of Alzheimer's disease by He et al.231 MicroRNA in tumor cells were detected by using exonuclease III and GO-regulated signal amplification by Huang et al.232 GO-capped mesoporous silica based drug could be released by remote control to cancer cells by photoinduced pH-jump activation as reported by He et al.233. Multifunctional hybrid nanopatches of GO and gold nanostars can be utilized for ultraefficient photothermal cancer therapy.234 The photothermal therapeutic response of cancer cells to aptamer–gold nanoparticle-hybridized GO under NIR illumination was studied by Yang et al.235 Multifunctional biocompatible GO quantum dots decorated magnetic nanoplatform were utilized for efficient capture and two-photon imaging of rare tumor cells.235
Demeritte et al.236 reported hybrid GO based plasmonic-magnetic multifunctional nanoplatform for selective separation and label-free identification of Alzheimer's disease biomarkers biotin containing rGO based nano system was proved as a multi-effect anticancer agent by Mauro et al.237 The hybrid based on rGO with ultrasmall gold nanorod were utilized for a sequential drug release and enhanced photothermal and photoacoustic effect for cancer therapy.238 Zirconia/GO hybrid micromotors can selectively capture the nerve agents as reported by Singh et al.239 Kenry et al.240 investigated the cellular interactions between the breast cancer cells and the two-dimensional planar graphene material films. They observed the selective accelerated proliferation and increased cellular spreading areas coupled with minimal cytotoxicity of both malignant breast cancer cells induced by the planar GO film.
GO–silver nanocomposite were reported as a highly effective antibacterial agent with species-specific mechanisms.241 This composite was also reported as a antibacterial against Xanthomonas perforans which causes a major disease of tomatoes, leading to reduction in production by 10–50%.242 GO wrapped SERS tags can also be utilized as a multifunctional platforms toward optical labeling, photothermal ablation of bacteria, and the monitoring of killing effect.243 Wang et al.244 synthesized the ZnO/GO composites of varying contents of ZnO in high quality and these composites own superior antibacterial properties against E. coli with low cytotoxicity. Besides GO utilized as anti-bacterial agent as a killing dental pathogens.245
5.5. Biosensors applications
Sensors/biosensors allow an electronic device to become a gateway between the digital and physical worlds. Sensor materials with unprecedented performance can create new applications and new avenues for user interaction. Graphene and its related materials have attracted much interest in sensing applications because of their optimized ratio between active surface and bulk volume. In particular, several forms of oxidized graphene have been studied to optimize the sensing efficiency, sometimes moving away from practical solutions to boost performance.246Biocompatible GO/functionalized GOs have been widely exploited for the sensing of biomolecules like (i) glucose,247 (ii) amplified DNA,248 (iii) D-glucosamine,249 (iv) microRNA,250 (v) DNA/RNA aptamers,251 (vi) multiplexed microRNA,252 (vii) optical aptamer,253 (viii) arbitrary DNA mutations,254 and (ix) luminescent iridium(III) complex labeled DNA.255 GO based CuO nanoparticles composite electrode highly enhanced the nonenzymatic glucose detection.256 A novel enzyme like activity of GO integrated with chitosan was demonstrated by Wang et al. and used as an efficient bio sensing system for glucose detection.257
5.6. Sensors applications
Ren et al.258 prepared GO/AgNP hybrids as SERS substrates for ultrasensitive and label-free SERS detection of folic acid in water and serum according to the inherent SERS spectra of folic acid. GO is also reported by various researchers as a ultrafast humidity sensors.259–261 Paek et al.262 developed a versatile platform for a highly stable, colorimetric, wide-range pH sensor. This pH sensor consisted of responsive polymer and QD hybrids integrated onto the surface of GO. The distances between the two color-emitting QDs and the GO were controlled independently via two linkers of pH-responsive poly(acrylic acid) (PAA) and poly(2-vinylpyridine) (P2VP), resulting in colorimetric responses over a wide range of pH values. Pt-decorated GO nanostructures were utilized by Wang et al. as a hydrogen gas sensor.2636. Conclusions and outlook
In this review we have intended to address the continued developments and challenges with a wide scope of interest, highlighting fundamental understanding of the synthesis, characterization, reduction of GO and its potential applications in various research fields. Graphene oxide and reduced graphene oxide (incomplete reduction into graphene due to the limited capacity of reducing agents), because of various containing functional groups like epoxy, hydroxyl, carboxyl and carbonyl, and various defects/disordered caused during reduction provide an efficient tool and a broad scope to scientific community for its widespread applications in emerging research areas of interest like energy sector and biomedicine. The GO is also a useful platform for the fabrication of functionalized graphene that can potentially confer improved mechanical, thermal and electronic properties etc. Readily available functional groups set up to bind with various other nanostructures, drugs and biological molecules like DNA, peptide, aptamer, protein, lipids etc. make graphene oxide as one of the most suitable candidate for energy storage, electrochemical devices, biosensors, catalysis, imaging/mapping of the cancer cells, photo-thermal therapy, targeted drug delivery, contamination purification and extraction devices for chemical, biological, and environmental samples. Various simple, large scale and cost effective methods discussed in the review have not only opened its application in the areas where we require large amount of materials but with reduced cost specially for drug design and delivery, the treatment/cure of life-threatening diseases like cancer and AIDS. Huge number of published research and works being done have not only proved its supremacy over other nanomaterials used so far but also continuously surprising researcher's mind with its new hope and possibilities day by day.Acknowledgements
Author D. P. Singh acknowledges with gratitude the financial support from CONICYT Chile (FONDECYT REGULAR 1151527).Notes and references
- J. Pedrós, A. Boscá, J. Martínez, S. Ruiz-Gómez, L. Pérez, V. Barranco and F. Calle, J. Power Sources, 2016, 317, 35–42 CrossRef.
- M. S. Dresselhaus, A. Jorio and R. Saito, Annu. Rev. Condens. Matter Phys., 2010, 1, 89–108 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- E. C. H. Sykes, Nat. Chem., 2009, 1, 175–176 CrossRef CAS PubMed.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170–1171 CrossRef CAS PubMed.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- S. V. Morozov, K. S. Novoselov, M. I. Katsnelson, F. Schedin, D. C. Elias, J. A. Jaszczak and A. K. Geim, Phys. Rev. Lett., 2008, 100, 016602 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 6217 CrossRef PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS.
- B. Marinho, M. Ghislandi, E. Tkalya, C. E. Koning and G. de With, Powder Technol., 2012, 221, 351–358 CrossRef CAS.
- A. J. Van Bommel, J. E. Crombeen and A. Van Tooren, Surf. Sci., 1975, 48, 463–472 CrossRef.
- H. P. Boehm, R. Setton and E. Stumpp, Carbon, 1986, 24, 241–245 CrossRef CAS.
- X. Lu, H. Huang, N. Nemchuk and R. S. Ruoff, Appl. Phys. Lett., 1999, 75, 193–195 CrossRef CAS.
- J. Wintterlin and M. L. Bocquet, Surf. Sci., 2009, 603, 1841–1852 CrossRef CAS.
- L. Xuekun, Y. Minfeng, H. Hui and S. R. Rodney, Nanotechnology, 1999, 10, 269 CrossRef.
- H. P. Boehm, A. Clauss, G. O. Fischer and U. Hofmann, Z. Anorg. Allg. Chem., 1962, 316, 119–127 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912–19916 CrossRef CAS.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1196 CrossRef CAS PubMed.
- T. A. Land, T. Michely, R. J. Behm, J. C. Hemminger and G. Comsa, Surf. Sci., 1992, 264, 261–270 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- M. Eizenberg and J. M. Blakely, Surf. Sci., 1979, 82, 228–236 CrossRef CAS.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249–259 CrossRef.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS.
- U. Hofmann and E. König, Z. Anorg. Allg. Chem., 1937, 234, 311–336 CrossRef CAS.
- Z. Liu, S. P. Lau and F. Yan, Chem. Soc. Rev., 2015, 44, 5638–5679 RSC.
- F. Perreault, A. Fonseca de Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC.
- K. Chen, S. Song, F. Liu and D. Xue, Chem. Soc. Rev., 2015, 44, 6230–6257 RSC.
- S. Guo and S. Dong, Chem. Soc. Rev., 2011, 40, 2644–2672 RSC.
- Y. Liu, X. Dong and P. Chen, Chem. Soc. Rev., 2012, 41, 2283–2307 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- R. Ruoff, Nat. Nanotechnol., 2008, 3, 10–11 CrossRef CAS PubMed.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS PubMed.
- F. Kim, L. J. Cote and J. Huang, Adv. Mater., 2010, 22, 1954–1958 CrossRef CAS PubMed.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538–542 CrossRef CAS PubMed.
- Y. Obeng and P. Srinivasan, Electrochem. Soc. Interface, 2011, 20, 47–52 CAS.
- C. Schafhaeutl, J. Prakt. Chem., 1840, 21, 129–157 CrossRef.
- C. Schafhaeutl, Philos. Mag., 1840, 16, 570–590 Search PubMed.
- U. Hofmann and R. Holst, Ber. Dtsch. Chem. Ges., 1939, 72, 754–771 CrossRef.
- C. Botas, P. Álvarez, P. Blanco, M. Granda, C. Blanco, R. Santamaría, L. J. Romasanta, R. Verdejo, M. A. López-Manchado and R. Menéndez, Carbon, 2013, 65, 156–164 CrossRef CAS.
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Boggild, S. Borini, F. H. L. Koppens, V. Palermo, N. Pugno, J. A. Garrido, R. Sordan, A. Bianco, L. Ballerini, M. Prato, E. Lidorikis, J. Kivioja, C. Marinelli, T. Ryhanen, A. Morpurgo, J. N. Coleman, V. Nicolosi, L. Colombo, A. Fert, M. Garcia-Hernandez, A. Bachtold, G. F. Schneider, F. Guinea, C. Dekker, M. Barbone, Z. Sun, C. Galiotis, A. N. Grigorenko, G. Konstantatos, A. Kis, M. Katsnelson, L. Vandersypen, A. Loiseau, V. Morandi, D. Neumaier, E. Treossi, V. Pellegrini, M. Polini, A. Tredicucci, G. M. Williams, B. Hee Hong, J.-H. Ahn, J. Min Kim, H. Zirath, B. J. van Wees, H. van der Zant, L. Occhipinti, A. Di Matteo, I. A. Kinloch, T. Seyller, E. Quesnel, X. Feng, K. Teo, N. Rupesinghe, P. Hakonen, S. R. T. Neil, Q. Tannock, T. Lofwander and J. Kinaret, Nanoscale, 2015, 7, 4598–4810 RSC.
- Q.-L. Yan, M. Gozin, F.-Q. Zhao, A. Cohen and S.-P. Pang, Nanoscale, 2016, 8, 4799–4851 RSC.
- T. Kuila, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Nanoscale, 2013, 5, 52–71 RSC.
- S. Mao, H. Pu and J. Chen, RSC Adv., 2012, 2, 2643–2662 RSC.
- H.-K. Jeong, Y. P. Lee, R. J. W. E. Lahaye, M.-H. Park, K. H. An, I. J. Kim, C.-W. Yang, C. Y. Park, R. S. Ruoff and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef CAS PubMed.
- R. Kumar, H.-J. Kim, S. Park, A. Srivastava and I.-K. Oh, Carbon, 2014, 79, 192–202 CrossRef CAS.
- R. Sun, H.-B. Zhang, J. Yao, D. Yang, Y.-W. Mai and Z.-Z. Yu, Carbon DOI:10.1016/j.carbon.2016.05.041.
- H. L. Poh, F. Šaněk, A. Ambrosi, G. Zhao, Z. Sofer and M. Pumera, Nanoscale, 2012, 4, 3515–3522 RSC.
- C. Hontoria-Lucas, A. J. López-Peinado, J. d. D. López-González, M. L. Rojas-Cervantes and R. M. Martín-Aranda, Carbon, 1995, 33, 1585–1592 CrossRef CAS.
- Y.-Y. Peng, Y.-M. Liu, J.-K. Chang, C.-H. Wu, M.-D. Ger, N.-W. Pu and C.-L. Chang, Carbon, 2015, 81, 347–356 CrossRef CAS.
- C. H. A. Wong, O. Jankovský, Z. Sofer and M. Pumera, Carbon, 2014, 77, 508–517 CrossRef CAS.
- H. L. Poh, P. Šimek, Z. Sofer and M. Pumera, ACS Nano, 2013, 7, 5262–5272 CrossRef CAS PubMed.
- D. Sačer, D. Čapeta, I. Šrut Rakić, R. Peter, M. Petravić and M. Kraljić Roković, Electrochim. Acta, 2016, 193, 311–320 CrossRef.
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- J. Chen, Y. Li, L. Huang, C. Li and G. Shi, Carbon, 2015, 81, 826–834 CrossRef CAS.
- J. Guerrero-Contreras and F. Caballero-Briones, Mater. Chem. Phys., 2015, 153, 209–220 CrossRef CAS.
- C.-I. Chang, K.-H. Chang, H.-H. Shen and C.-C. Hu, J. Taiwan Inst. Chem. Eng., 2014, 45, 2762–2769 CrossRef CAS.
- J. H. Kang, T. Kim, J. Choi, J. Park, Y. S. Kim, M. S. Chang, H. Jung, K. T. Park, S. J. Yang and C. R. Park, Chem. Mater., 2016, 28, 756–764 CrossRef CAS.
- U. Saha, R. Jaiswal and T. H. Goswami, Electrochim. Acta, 2016, 196, 386–404 CrossRef CAS.
- D. Pacilé, J. C. Meyer, A. Fraile Rodríguez, M. Papagno, C. Gómez-Navarro, R. S. Sundaram, M. Burghard, K. Kern, C. Carbone and U. Kaiser, Carbon, 2011, 49, 966–972 CrossRef.
- J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. Ye, Chem. Mater., 2009, 21, 3514–3520 CrossRef CAS.
- M. Wissler, J. Power Sources, 2006, 156, 142–150 CrossRef CAS.
- A. Dimiev, D. V. Kosynkin, L. B. Alemany, P. Chaguine and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CAS.
- W. Zhang, V. Carravetta, Z. Li, Y. Luo and J. Yang, J. Chem. Phys., 2009, 131, 244505 CrossRef PubMed.
- D. W. Lee, L. V. De Los Santos, J. W. Seo, L. L. Felix, A. Bustamante, D. J. M. Cole and C. H. W. Barnes, J. Phys. Chem. B, 2010, 114, 5723–5728 CrossRef CAS PubMed.
- J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2009, 25, 5957–5968 CrossRef CAS PubMed.
- S. Saxena, T. A. Tyson and E. Negusse, J. Phys. Chem. Lett., 2010, 1, 3433–3437 CrossRef CAS.
- V. Lee, L. Whittaker, C. Jaye, K. M. Baroudi, D. A. Fischer and S. Banerjee, Chem. Mater., 2009, 21, 3905–3916 CrossRef CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Phys. Chem. C, 2011, 115, 19761–19781 CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
- L. B. Casabianca, M. A. Shaibat, W. W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 5672–5676 CrossRef CAS PubMed.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice Jr and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- O. Akhavan, Carbon, 2010, 48, 509–519 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- B. Sun, X. Huang, S. Chen, P. Munroe and G. Wang, Nano Lett., 2014, 14, 3145–3152 CrossRef CAS PubMed.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1290 RSC.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- H.-J. Shin, K. K. Kim, A. Benayad, S.-M. Yoon, H. K. Park, I.-S. Jung, M. H. Jin, H.-K. Jeong, J. M. Kim, J.-Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- V. H. Pham, T. V. Cuong, T.-D. Nguyen-Phan, H. D. Pham, E. J. Kim, S. H. Hur, E. W. Shin, S. Kim and J. S. Chung, Chem. Commun., 2010, 46, 4375–4377 RSC.
- T. Cassagneau and J. H. Fendler, J. Phys. Chem. B, 1999, 103, 1789–1793 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 Search PubMed.
- Z. Li, Y. Yao, Z. Lin, K.-S. Moon, W. Lin and C. Wong, J. Mater. Chem., 2010, 20, 4781–4783 RSC.
- X. Zhou, J. Zhang, H. Wu, H. Yang, J. Zhang and S. Guo, J. Phys. Chem. C, 2011, 115, 11957–11961 CAS.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CAS.
- C. A. Amarnath, C. E. Hong, N. H. Kim, B.-C. Ku, T. Kuila and J. H. Lee, Carbon, 2011, 49, 3497–3502 CrossRef CAS.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- T. A. Pham, J. S. Kim, J. S. Kim and Y. T. Jeong, Colloids Surf., A, 2011, 384, 543–548 CrossRef CAS.
- Z. Lei, L. Lu and X. S. Zhao, Energy Environ. Sci., 2012, 5, 6391–6399 CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- Z.-S. Wu, W. Ren, L. Gao, J. Zhao, Z. Chen, B. Liu, D. Tang, B. Yu, C. Jiang and H.-M. Cheng, ACS Nano, 2009, 3, 411–417 CrossRef CAS PubMed.
- G. I. Titelman, V. Gelman, S. Bron, R. L. Khalfin, Y. Cohen and H. Bianco-Peled, Carbon, 2005, 43, 641–649 CrossRef CAS.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- A. M. Schwenke, S. Hoeppener and U. S. Schubert, J. Mater. Chem. A, 2015, 3, 23778–23787 CAS.
- Y. Zhu, S. Murali, M. D. Stoller, A. Velamakanni, R. D. Piner and R. S. Ruoff, Carbon, 2010, 48, 2118–2122 CrossRef CAS.
- H. M. A. Hassan, V. Abdelsayed, A. E. R. S. Khder, K. M. AbouZeid, J. Terner, M. S. El-Shall, S. I. Al-Resayes and A. A. El-Azhary, J. Mater. Chem., 2009, 19, 3832–3837 RSC.
- Y. Zhang, L. Guo, S. Wei, Y. He, H. Xia, Q. Chen, H.-B. Sun and F.-S. Xiao, Nano Today, 2010, 5, 15–20 CrossRef CAS.
- L. J. Cote, R. Cruz-Silva and J. Huang, J. Am. Chem. Soc., 2009, 131, 11027–11032 CrossRef CAS PubMed.
- T. V. Khai, D. S. Kwak, Y. J. Kwon, H. Y. Cho, T. N. Huan, H. Chung, H. Ham, C. Lee, N. V. Dan, N. T. Tung and H. W. Kim, Chem. Eng. J., 2013, 232, 346–355 CrossRef CAS.
- H. J. Han, Y. N. Chen and Z. J. Wang, RSC Adv., 2015, 5, 92940–92946 RSC.
- T. Lu, L. Pan, C. Nie, Z. Zhao and Z. Sun, Phys. Status Solidi A, 2011, 208, 2325–2327 CrossRef CAS.
- D. Sun, L. Jin, Y. Chen, J.-R. Zhang and J.-J. Zhu, ChemPlusChem, 2013, 78, 227–234 CrossRef CAS.
- A. Y. S. Eng, Z. Sofer, P. Šimek, J. Kosina and M. Pumera, Chem.–Eur. J., 2013, 19, 15583–15592 CrossRef CAS PubMed.
- A. M. Schwenke, S. Hoeppener and U. S. Schubert, Adv. Mater., 2015, 27, 4113–4141 CrossRef CAS PubMed.
- Q. Yan, Q. Liu and J. Wang, Ceram. Int., 2016, 42, 3007–3013 CrossRef CAS.
- W. Chen, L. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS.
- C. N. R. Rao, K. S. Subrahmanyam, H. S. S. R. Matte, B. Abdulhakeem, A. Govindaraj, D. Barun, K. Prashant, G. Anupama and J. L. Dattatray, Sci. Technol. Adv. Mater., 2010, 11, 054502 CrossRef.
- Y. H. Ding, P. Zhang, Q. Zhuo, H. M. Ren, Z. M. Yang and Y. Jiang, Nanotechnology, 2011, 22, 215601 CrossRef CAS PubMed.
- V. Abdelsayed, S. Moussa, H. M. Hassan, H. S. Aluri, M. M. Collinson and M. S. El-Shall, J. Phys. Chem. Lett., 2010, 1, 2804–2809 CrossRef CAS.
- P. Kumar, B. Das, B. Chitara, K. S. Subrahmanyam, K. Gopalakrishnan, S. B. Krupanidhi and C. N. R. Rao, Macromol. Chem. Phys., 2012, 213, 1146–1163 CrossRef CAS.
- P. Kumar, K. S. Subrahmanyam and C. N. R. Rao, Mater. Express, 2011, 1, 252–256 CrossRef CAS.
- M. Baraket, S. G. Walton, Z. Wei, E. H. Lock, J. T. Robinson and P. Sheehan, Carbon, 2010, 48, 3382–3390 CrossRef CAS.
- O. Ö. Ekiz, M. Ürel, H. Güner, A. K. Mizrak and A. Dâna, ACS Nano, 2011, 5, 2475–2482 CrossRef CAS PubMed.
- P. Yao, P. Chen, L. Jiang, H. Zhao, H. Zhu, D. Zhou, W. Hu, B.-H. Han and M. Liu, Adv. Mater., 2010, 22, 5008–5012 CrossRef CAS PubMed.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS PubMed.
- P. V. Kamat, Chem. Rev., 1993, 93, 267–300 CrossRef CAS.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- S. R. Kim, M. K. Parvez and M. Chhowalla, Chem. Phys. Lett., 2009, 483, 124–127 CrossRef CAS.
- N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed.
- A. Kongkanand and P. V. Kamat, ACS Nano, 2007, 1, 13–21 CrossRef CAS PubMed.
- P. V. Kamat, I. Bedja and S. Hotchandani, J. Phys. Chem., 1994, 98, 9137–9142 CrossRef CAS.
- G. Williams and P. V. Kamat, Langmuir, 2009, 25, 13869–13873 CrossRef CAS PubMed.
- S. Dubin, S. Gilje, K. Wang, V. C. Tung, K. Cha, A. S. Hall, J. Farrar, R. Varshneya, Y. Yang and R. B. Kaner, ACS Nano, 2010, 4, 3845–3852 CrossRef CAS PubMed.
- H. Wang, J. T. Robinson, X. Li and H. Dai, J. Am. Chem. Soc., 2009, 131, 9910–9911 CrossRef CAS PubMed.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- G. Demazeau, J. Mater. Chem., 1999, 9, 15–18 RSC.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- L.-B. Luo, S.-H. Yu, H.-S. Qian and T. Zhou, J. Am. Chem. Soc., 2005, 127, 2822–2823 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- X. Shen, L. Jiang, Z. Ji, J. Wu, H. Zhou and G. Zhu, J. Colloid Interface Sci., 2011, 354, 493–497 CrossRef CAS PubMed.
- W. Chen, L. Yan and P. R. Bangal, J. Phys. Chem. C, 2010, 114, 19885–19890 CAS.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS PubMed.
- Z.-J. Fan, W. Kai, J. Yan, T. Wei, L.-J. Zhi, J. Feng, Y.-M. Ren, L.-P. Song and F. Wei, ACS Nano, 2011, 5, 191–198 CrossRef CAS PubMed.
- R. S. Dey, S. Hajra, R. K. Sahu, C. R. Raj and M. K. Panigrahi, Chem. Commun., 2012, 48, 1787–1789 RSC.
- T. S. Sreeprasad, A. K. Samal and T. Pradeep, J. Phys. Chem. C, 2009, 113, 1727–1737 CAS.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS.
- W. Chen and L. Yan, Nanoscale, 2010, 2, 559–563 RSC.
- X. Zhu, Y. Zhu, S. Murali, M. D. Stoller and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS PubMed.
- F. Zhang, H. Cao, D. Yue, J. Zhang and M. Qu, Inorg. Chem., 2012, 51, 9544–9551 CrossRef CAS PubMed.
- B. Wang, X. Li, X. Zhang, B. Luo, M. Jin, M. Liang, S. A. Dayeh, S. T. Picraux and L. Zhi, ACS Nano, 2013, 7, 1437–1445 CrossRef CAS PubMed.
- A. Bhaskar, M. Deepa, M. Ramakrishna and T. N. Rao, J. Phys. Chem. C, 2014, 118, 7296–7306 CAS.
- X. Guan, J. Nai, Y. Zhang, P. Wang, J. Yang, L. Zheng, J. Zhang and L. Guo, Chem. Mater., 2014, 26, 5958–5964 CrossRef CAS.
- J. Yao, Y. Gong, S. Yang, P. Xiao, Y. Zhang, K. Keyshar, G. Ye, S. Ozden, R. Vajtai and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2014, 6, 20414–20422 CAS.
- Y. Xu, X. Zhu, X. Zhou, X. Liu, Y. Liu, Z. Dai and J. Bao, J. Phys. Chem. C, 2014, 118, 28502–28508 CAS.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- D. Yoon, K. Y. Chung, W. Chang, S. M. Kim, M. J. Lee, Z. Lee and J. Kim, Chem. Mater., 2015, 27, 266–275 CrossRef CAS.
- X. Liu, J. Zhang, W. Si, L. Xi, B. Eichler, C. Yan and O. G. Schmidt, ACS Nano, 2015, 9, 1198–1205 CrossRef CAS PubMed.
- G. Zeng, N. Shi, M. Hess, X. Chen, W. Cheng, T. Fan and M. Niederberger, ACS Nano, 2015, 9, 4227–4235 CrossRef CAS PubMed.
- X. Gao, J. Li, Y. Xie, D. Guan and C. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 7855–7862 CAS.
- F. Xiong, Z. Cai, L. Qu, P. Zhang, Z. Yuan, O. K. Asare, W. Xu, C. Lin and L. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 12625–12630 CAS.
- S. H. Choi, K. Y. Jung and Y. C. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 13952–13959 CAS.
- C. Feng, L. Zhang, M. Yang, X. Song, H. Zhao, Z. Jia, K. Sun and G. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 15726–15734 CAS.
- A. R. Park, D.-Y. Son, J. S. Kim, J. Y. Lee, N.-G. Park, J. Park, J. K. Lee and P. J. Yoo, ACS Appl. Mater. Interfaces, 2015, 7, 18483–18490 CAS.
- X. Xu, S. Ji, M. Gu and J. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 20957–20964 CAS.
- X. Wei, C. Tang, X. Wang, L. Zhou, Q. Wei, M. Yan, J. Sheng, P. Hu, B. Wang and L. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 26572–26578 CAS.
- H. Xu, L. Shi, Z. Wang, J. Liu, J. Zhu, Y. Zhao, M. Zhang and S. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 27486–27493 CAS.
- K. S. Lee, S. Park, W. Lee and Y. S. Yoon, ACS Appl. Mater. Interfaces, 2016, 8, 2027–2034 CAS.
- Y. Luo, X. Xu, Y. Zhang, C.-Y. Chen, L. Zhou, M. Yan, Q. Wei, X. Tian and L. Mai, ACS Appl. Mater. Interfaces, 2016, 8, 2812–2818 CAS.
- S. H. Ha, Y. S. Jeong and Y. J. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 12295–12303 CAS.
- Z. Guo, D. Zhang, H. Qiu, T. Zhang, Q. Fu, L. Zhang, X. Yan, X. Meng, G. Chen and Y. Wei, ACS Appl. Mater. Interfaces, 2015, 7, 13972–13979 CAS.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- W. Zhou, H. Chen, Y. Yu, D. Wang, Z. Cui, F. J. DiSalvo and H. D. Abruña, ACS Nano, 2013, 7, 8801–8808 CrossRef CAS PubMed.
- J. Rong, M. Ge, X. Fang and C. Zhou, Nano Lett., 2014, 14, 473–479 CrossRef CAS PubMed.
- C. Wang, X. Wang, Y. Yang, A. Kushima, J. Chen, Y. Huang and J. Li, Nano Lett., 2015, 15, 1796–1802 CrossRef CAS PubMed.
- Y. Hwa, J. Zhao and E. J. Cairns, Nano Lett., 2015, 15, 3479–3486 CrossRef CAS PubMed.
- J.-Q. Huang, T.-Z. Zhuang, Q. Zhang, H.-J. Peng, C.-M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS PubMed.
- H.-G. Jung, Y. S. Jeong, J.-B. Park, Y.-K. Sun, B. Scrosati and Y. J. Lee, ACS Nano, 2013, 7, 3532–3539 CrossRef CAS PubMed.
- R. Black, J.-H. Lee, B. Adams, C. A. Mims and L. F. Nazar, Angew. Chem., 2013, 125, 410–414 CrossRef.
- Y. Li, J. Wang, X. Li, D. Geng, R. Li and X. Sun, Chem. Commun., 2011, 47, 9438–9440 RSC.
- Y. Li, J. Wang, X. Li, D. Geng, M. N. Banis, Y. Tang, D. Wang, R. Li, T.-K. Sham and X. Sun, J. Mater. Chem., 2012, 22, 20170–20174 RSC.
- Y. Li, J. Wang, X. Li, D. Geng, M. N. Banis, R. Li and X. Sun, Electrochem. Commun., 2012, 18, 12–15 CrossRef CAS.
- S.-S. Li, K.-H. Tu, C.-C. Lin, C.-W. Chen and M. Chhowalla, ACS Nano, 2010, 4, 3169–3174 CrossRef CAS PubMed.
- J. Liu, Y. Xue and L. Dai, J. Phys. Chem. Lett., 2012, 3, 1928–1933 CrossRef CAS PubMed.
- J. G. Radich, R. Dwyer and P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 2453–2460 CrossRef CAS.
- I. P. Murray, S. J. Lou, L. J. Cote, S. Loser, C. J. Kadleck, T. Xu, J. M. Szarko, B. S. Rolczynski, J. E. Johns, J. Huang, L. Yu, L. X. Chen, T. J. Marks and M. C. Hersam, J. Phys. Chem. Lett., 2011, 2, 3006–3012 CrossRef CAS.
- L. Kavan, J.-H. Yum and M. Graetzel, ACS Appl. Mater. Interfaces, 2012, 4, 6999–7006 CAS.
- Y. Li, H. Wang, Q. Feng, G. Zhou and Z.-S. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 8217–8224 CAS.
- M.-H. Yeh, L.-Y. Lin, C.-L. Sun, Y.-A. Leu, J.-T. Tsai, C.-Y. Yeh, R. Vittal and K.-C. Ho, J. Phys. Chem. C, 2014, 118, 16626–16634 CAS.
- Z. Xiang, X. Zhou, G. Wan, G. Zhang and D. Cao, ACS Sustainable Chem. Eng., 2014, 2, 1234–1240 CrossRef CAS.
- H.-W. Liu, S.-P. Liang, T.-J. Wu, H. Chang, P.-K. Kao, C.-C. Hsu, J.-Z. Chen, P.-T. Chou and I. C. Cheng, ACS Appl. Mater. Interfaces, 2014, 6, 15105–15112 CAS.
- B. Yang, X. Zuo, P. Chen, L. Zhou, X. Yang, H. Zhang, G. Li, M. Wu, Y. Ma, S. Jin and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 137–143 CAS.
- B. Chen, J. Sha, W. Li, F. He, E. Liu, C. Shi, C. He, J. Li and N. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 2495–2504 CAS.
- G. S. Han, Y. H. Song, Y. U. Jin, J.-W. Lee, N.-G. Park, B. K. Kang, J.-K. Lee, I. S. Cho, D. H. Yoon and H. S. Jung, ACS Appl. Mater. Interfaces, 2015, 7, 23521–23526 CAS.
- N. A. Kumar, H.-J. Choi, Y. R. Shin, D. W. Chang, L. Dai and J.-B. Baek, ACS Nano, 2012, 6, 1715–1723 CrossRef CAS PubMed.
- G. Xu, N. Wang, J. Wei, L. Lv, J. Zhang, Z. Chen and Q. Xu, Ind. Eng. Chem. Res., 2012, 51, 14390–14398 CrossRef CAS.
- D. Xu, Q. Xu, K. Wang, J. Chen and Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 200–209 CAS.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS PubMed.
- W. Li, Y. Bu, H. Jin, J. Wang, W. Zhang, S. Wang and J. Wang, Energy Fuels, 2013, 27, 6304–6310 CrossRef CAS.
- C.-T. Hsieh, W.-Y. Lee, C.-E. Lee and H. Teng, J. Phys. Chem. C, 2014, 118, 15146–15153 CAS.
- L.-B. Xing, S.-F. Hou, J. Zhou, S. Li, T. Zhu, Z. Li, W. Si and S. Zhuo, J. Phys. Chem. C, 2014, 118, 25924–25930 CAS.
- V. Sahu, S. Shekhar, R. K. Sharma and G. Singh, ACS Appl. Mater. Interfaces, 2015, 7, 3110–3116 CAS.
- S. Cho, J. S. Lee and J. Jang, ACS Appl. Mater. Interfaces, 2015, 7, 9668–9681 CAS.
- R. Kumar, R. K. Singh, P. K. Dubey, D. P. Singh and R. M. Yadav, ACS Appl. Mater. Interfaces, 2015, 7, 15042–15051 CAS.
- K. Jo, M. Gu and B.-S. Kim, Chem. Mater., 2015, 27, 7982–7989 CrossRef CAS.
- J. Yang, E. Zhang, X. Li, Y. Yu, J. Qu and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 2297–2305 CAS.
- M. Wu, R. Kempaiah, P.-J. J. Huang, V. Maheshwari and J. Liu, Langmuir, 2011, 27, 2731–2738 CrossRef CAS PubMed.
- J. S. Park, N.-I. Goo and D.-E. Kim, Langmuir, 2014, 30, 12587–12595 CrossRef CAS PubMed.
- H. Zhang, C. Peng, J. Yang, M. Lv, R. Liu, D. He, C. Fan and Q. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 1761–1767 CAS.
- G. Liu, H. Shen, J. Mao, L. Zhang, Z. Jiang, T. Sun, Q. Lan and Z. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 6909–6914 CAS.
- M. Zhang, Y. Cao, Y. Chong, Y. Ma, H. Zhang, Z. Deng, C. Hu and Z. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 13325–13332 CAS.
- C. L. Weaver, J. M. LaRosa, X. Luo and X. T. Cui, ACS Nano, 2014, 8, 1834–1843 CrossRef CAS PubMed.
- K. Li, L. Feng, J. Shen, Q. Zhang, Z. Liu, S.-T. Lee and J. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 5900–5907 CAS.
- A. Paul, A. Hasan, H. A. Kindi, A. K. Gaharwar, V. T. S. Rao, M. Nikkhah, S. R. Shin, D. Krafft, M. R. Dokmeci, D. Shum-Tim and A. Khademhosseini, ACS Nano, 2014, 8, 8050–8062 CrossRef CAS PubMed.
- E. Song, W. Han, C. Li, D. Cheng, L. Li, L. Liu, G. Zhu, Y. Song and W. Tan, ACS Appl. Mater. Interfaces, 2014, 6, 11882–11890 CAS.
- X. Zhao, L. Yang, X. Li, X. Jia, L. Liu, J. Zeng, J. Guo and P. Liu, Bioconjugate Chem., 2015, 26, 128–136 CrossRef CAS PubMed.
- H. Kim, R. Namgung, K. Singha, I.-K. Oh and W. J. Kim, Bioconjugate Chem., 2011, 22, 2558–2567 CrossRef CAS PubMed.
- C. Xu, D. Yang, L. Mei, B. Lu, L. Chen, Q. Li, H. Zhu and T. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 2715–2724 CAS.
- L.-N. Zhang, H.-H. Deng, F.-L. Lin, X.-W. Xu, S.-H. Weng, A.-L. Liu, X.-H. Lin, X.-H. Xia and W. Chen, Anal. Chem., 2014, 86, 2711–2718 CrossRef CAS PubMed.
- X.-P. He, Q. Deng, L. Cai, C.-Z. Wang, Y. Zang, J. Li, G.-R. Chen and H. Tian, ACS Appl. Mater. Interfaces, 2014, 6, 5379–5382 CAS.
- R.-C. Huang, W.-J. Chiu, Y.-J. Li and C.-C. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 21780–21787 CAS.
- D. He, X. He, K. Wang, Z. Zou, X. Yang and X. Li, Langmuir, 2014, 30, 7182–7189 CrossRef CAS PubMed.
- S. Z. Nergiz, N. Gandra, S. Tadepalli and S. Singamaneni, ACS Appl. Mater. Interfaces, 2014, 6, 16395–16402 CAS.
- L. Yang, Y.-T. Tseng, G. Suo, L. Chen, J. Yu, W.-J. Chiu, C.-C. Huang and C.-H. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 5097–5106 CAS.
- T. Demeritte, B. P. Viraka Nellore, R. Kanchanapally, S. S. Sinha, A. Pramanik, S. R. Chavva and P. C. Ray, ACS Appl. Mater. Interfaces, 2015, 7, 13693–13700 CAS.
- N. Mauro, C. Scialabba, G. Cavallaro, M. Licciardi and G. Giammona, Biomacromolecules, 2015, 16, 2766–2775 CrossRef CAS PubMed.
- J. Song, X. Yang, O. Jacobson, L. Lin, P. Huang, G. Niu, Q. Ma and X. Chen, ACS Nano, 2015, 9, 9199–9209 CrossRef CAS PubMed.
- V. V. Singh, A. Martin, K. Kaufmann, S. D. S. de Oliveira and J. Wang, Chem. Mater., 2015, 27, 8162–8169 CrossRef CAS.
- Kenry, P. K. Chaudhuri, K. P. Loh and C. T. Lim, ACS Nano, 2016, 10, 3424–3434 CrossRef CAS PubMed.
- J. Tang, Q. Chen, L. Xu, S. Zhang, L. Feng, L. Cheng, H. Xu, Z. Liu and R. Peng, ACS Appl. Mater. Interfaces, 2013, 5, 3867–3874 CAS.
- I. Ocsoy, M. L. Paret, M. A. Ocsoy, S. Kunwar, T. Chen, M. You and W. Tan, ACS Nano, 2013, 7, 8972–8980 CrossRef CAS PubMed.
- D. Lin, T. Qin, Y. Wang, X. Sun and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 1320–1329 CAS.
- Y.-W. Wang, A. Cao, Y. Jiang, X. Zhang, J.-H. Liu, Y. Liu and H. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 2791–2798 CAS.
- J. He, X. Zhu, Z. Qi, C. Wang, X. Mao, C. Zhu, Z. He, M. Li and Z. Tang, ACS Appl. Mater. Interfaces, 2015, 7, 5605–5611 CAS.
- S. Prezioso, F. Perrozzi, L. Giancaterini, C. Cantalini, E. Treossi, V. Palermo, M. Nardone, S. Santucci and L. Ottaviano, J. Phys. Chem. C, 2013, 117, 10683–10690 CAS.
- Y. Liu, D. Yu, C. Zeng, Z. Miao and L. Dai, Langmuir, 2010, 26, 6158–6160 CrossRef CAS PubMed.
- X. Liu, R. Aizen, R. Freeman, O. Yehezkeli and I. Willner, ACS Nano, 2012, 6, 3553–3563 CrossRef CAS PubMed.
- R. Cheng, Y. Liu, S. Ou, Y. Pan, S. Zhang, H. Chen, L. Dai and J. Qu, Anal. Chem., 2012, 84, 5641–5644 CrossRef CAS PubMed.
- Y. Tu, W. Li, P. Wu, H. Zhang and C. Cai, Anal. Chem., 2013, 85, 2536–2542 CrossRef CAS PubMed.
- Y. Wang, Z. Li, T. J. Weber, D. Hu, C.-T. Lin, J. Li and Y. Lin, Anal. Chem., 2013, 85, 6775–6782 CrossRef CAS PubMed.
- S.-R. Ryoo, J. Lee, J. Yeo, H.-K. Na, Y.-K. Kim, H. Jang, J. H. Lee, S. W. Han, Y. Lee, V. N. Kim and D.-H. Min, ACS Nano, 2013, 7, 5882–5891 CrossRef CAS PubMed.
- X. Liu, F. Wang, R. Aizen, O. Yehezkeli and I. Willner, J. Am. Chem. Soc., 2013, 135, 11832–11839 CrossRef CAS PubMed.
- J. Huang, Z. Wang, J.-K. Kim, X. Su and Z. Li, Anal. Chem., 2015, 87, 12254–12261 CrossRef CAS PubMed.
- Q. Zhao, Y. Zhou, Y. Li, W. Gu, Q. Zhang and J. Liu, Anal. Chem., 2016, 88, 1892–1899 CrossRef CAS PubMed.
- J. Song, L. Xu, C. Zhou, R. Xing, Q. Dai, D. Liu and H. Song, ACS Appl. Mater. Interfaces, 2013, 5, 12928–12934 CAS.
- G.-L. Wang, X. Xu, X. Wu, G. Cao, Y. Dong and Z. Li, J. Phys. Chem. C, 2014, 118, 28109–28117 CAS.
- W. Ren, Y. Fang and E. Wang, ACS Nano, 2011, 5, 6425–6433 CrossRef CAS PubMed.
- S. Borini, R. White, D. Wei, M. Astley, S. Haque, E. Spigone, N. Harris, J. Kivioja and T. Ryhänen, ACS Nano, 2013, 7, 11166–11173 CrossRef CAS PubMed.
- S.-H. Hwang, D. Kang, R. S. Ruoff, H. S. Shin and Y.-B. Park, ACS Nano, 2014, 8, 6739–6747 CrossRef CAS PubMed.
- H.-W. Yu, H. K. Kim, T. Kim, K. M. Bae, S. M. Seo, J.-M. Kim, T. J. Kang and Y. H. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 8320–8326 CAS.
- K. Paek, H. Yang, J. Lee, J. Park and B. J. Kim, ACS Nano, 2014, 8, 2848–2856 CrossRef CAS PubMed.
- J. Wang, S. Rathi, B. Singh, I. Lee, H.-I. Joh and G.-H. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 13768–13775 CAS.
| This journal is © The Royal Society of Chemistry 2016 |