
New design of highly homogeneous photopolymer networks for shape memory materials†
Matthieu Retailleau,
Ahmad Ibrahim,
Céline Croutxé-Barghorn* and
Xavier Allonas
Laboratory of Macromolecular Photochemistry and Engineering, University of Haute Alsace, 3b rue Alfred Werner, 68093 Mulhouse, France. E-mail: celine.croutxe-barghorn@uha.fr
First published on 4th May 2016
Abstract
A shape memory polymer (SMP) based on diacrylates and primary diamines was designed by ingeniously sequencing double click aza-Michael addition and radical photopolymerization. The chemical approach lies in an additional crosslinking of 2-step based poly(amino ester)–poly(acrylate)s thus, resulting in enhanced polymer network homogeneity and thermomechanical properties. Photopolymers exhibiting excellent shape fixity and recovery within a narrow temperature range were achieved.
Shape memory polymers (SMP) are materials that are able to undergo programmable deformation. After being first moulded in their permanent form, the polymer can be deformed and fixed in a temporary form. Then, the polymer can return to its original form upon application of an external stimulus (heat, light, water immersion…).1–5 Generally, the most used external stimulus is heat as polymer transition temperature (Ttrans) can easily be tuned.6,8 As a consequence, the polymer should be highly responsive to thermal changes meaning that the polymerization process should end up with a uniform distribution of functional groups. In brief, the higher the polymer network homogeneity is, the better the shape memory performances are.5
With a view to develop scientific, industrial and commercial applications, many chemistries have been proposed for SMP.4 Among them, (meth)acrylate based materials have been investigated via photopolymerization process.7–9 Indeed, photochemical processes afford a spatial and temporal control of the polymerization process while being fast.10 However, chain growth mechanism generally involved in free radical photopolymerization result in the formation of heterogeneous polymer networks with broad temperature range for the glassy to rubbery transition and high shrinkage.10 In order to improve the network homogeneity and reduce shrinkage, SMP based on thiol click photochemistry have been reported in the literature.11 This approach presents the advantages of producing homogeneous network with narrow glass transition temperature, leading to high fixation rates and fast recovery rates (3% °C−1).11,12 Nevertheless, it ends up with remaining dangling chains that are considered as network defects since they do not contribute to the overall structure of the polymer network. In that sense, they are responsible for network heterogeneity and decreased SMP properties.
Herein, we describe an alternative approach based on a three-step photopolymerization system for a new SMP generation. Our strategy consists in combining double click aza-Michael addition and radical photopolymerization13 (Scheme 1). In a first step, a blend of linear polymers in presence of unreacted diacrylates monomers results from click-reactions between bisphenol A ethoxylated diacrylates (which are in excess) and pentane-1,5-diamines (AZ1). Then, remaining unreacted diacrylate groups are photopolymerized to increase mechanical properties of the network during a second step (hν). In a third step, the trapped acrylate dangling groups react via a second click reaction (AZ2) with the secondary amines resulting from the first step. Thus, AZ2 reaction reinforces the network homogeneity, enhancing thermomechanical properties of the final polymer.13
 | ||
| Scheme 1 Network evolution via a three step polymerization using double click aza-Michael addition and radical photopolymerization. | ||
The whole process combines advantageous properties (low shrinkage, tunable properties…) as for the thiol click system11 without facing the presence of unreactive nucleophile groups in the final network. Starting from different amine/acrylate ratios, the influence of AZ2 on the network homogeneity was first underlined without any involvement of photopolymerization step. Then, a polymer network formed within a three-step process including a photopolymerization step was formed and its potential in terms of shape memory polymer was investigated. The effect of the AZ2 third step was correlated to SMPs characteristics.
The effectiveness of AZ2 reaction was first explored by DSC without any photopolymerization step for different amine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acrylate ratios (see ESI for more details about materials and methods†). Slope of the glass transition curve and ΔTg are relevant data to assess AZ2 contribution with respect to the AZ1 one. Indeed, a sharp transition and a narrow temperature range suggest usually a homogeneous polymer network.14 Different trends depending on the ratio studied can be observed in Table 1. 1:2 ratio is the most impacted by AZ2 (ΔTg broadens by 10 °C and the slope decreases by 70%). Regarding 1:4 ratio, ΔTg widens by 5 °C and the slope diminishes by 45%.
:acrylate ratios (see ESI for more details about materials and methods†). Slope of the glass transition curve and ΔTg are relevant data to assess AZ2 contribution with respect to the AZ1 one. Indeed, a sharp transition and a narrow temperature range suggest usually a homogeneous polymer network.14 Different trends depending on the ratio studied can be observed in Table 1. 1:2 ratio is the most impacted by AZ2 (ΔTg broadens by 10 °C and the slope decreases by 70%). Regarding 1:4 ratio, ΔTg widens by 5 °C and the slope diminishes by 45%.
:2, 1:4 and 1:8 (ΔTg = To − Te, where To is onset and Te is the end of transition; slope: slope of the glass transition curve). Derivative curves of heat flow are reported in ESI
NH2:C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
End of AZ1 | End of AZ2 | ||||
|---|---|---|---|---|---|---|
| Tg | Slope | ΔTg | Tg | Slope | ΔTg | |
| 1:2 |
−23 | −0.020 | 5 | 6 | −0.006 | 15 |
| 1:4 |
−31 | −0.022 | 5 | −11 | −0.012 | 10 |
| 1:8 |
−34 | −0.026 | 4 | −28 | −0.023 | 5 |
As reported in the literature,15 crosslinking of the linear polymer during AZ2 occurs in a random manner and results in polymeric network having a rather undefined network structure. By contrast, 1:8 ratio leads to polymer with the lowest Tg variation between AZ1 and AZ2 and the lowest Tg after AZ1 + AZ2 completion. ΔTg remains unchanged and the slope barely changes. Presence of higher amount of unreacted acrylates which are not taking part in AZ2 accounts for these results.
Turning now to the three-step process (i.e. AZ1 + hν + AZ2), it is expected that a photoinduced radical polymerization of acrylates after AZ1 would raise the whole polymer conversion. Additionally, AZ2 would consume residual double-bonds and improve the properties of the final network. Thus, it is of prime interest to evaluate the contribution of AZ2 in this three-step AZ1 + hν + AZ2 approach (using 2.5 wt% of Irgacure 819 as photoinitiator).
For this study, 1:2 ratio was not considered. Indeed, although a polymer network with high thermomechanical properties could be achieved in this case, the important viscosity of the mixture reached right after AZ1 does not enable the formation a thin film that can be further photopolymerized and characterized. 1:8 ratio was not selected because of the limited impact of AZ2 on final Tg. As a consequence, 1:4 ratio appears as the best candidate to investigate the potential of the three-step based polymer as a thermally-triggered SMP. As AZ2 proceeds over couple of days, the thermomechanical properties of 1:4 ratio were characterized as a function of corresponding ageing time (Table 2). Interestingly, the rubbery modulus increased from 15.3 (t = 0 day) to 27 MPa (t = 25 day) while the glassy modulus remains relatively constant within experimental error. As known, the change in rubbery modulus is driven by an increase in crosslink density.15 Therefore, the molecular weight between crosslinks dropped and Mc dispersity is reduced during AZ2 reaction. In addition, the shortening of ΔTg and tan(δ) peak width at half its height indicates an improvement of network homogeneity. Thus, the decrease in tan(δ) amplitude can be attributed to the combination of higher crosslink density and constrained chain mobility.16,17 Consequently, by clicking acrylate dangling groups, AZ2 reaction increases the crosslink density and results in an increased network homogeneity as observed in other studies.18 The occurrence of AZ2 reaction in the three-step AZ1 + hν + AZ2 process is a clear benefit for the synthesis of highly homogeneous polymer network. These results should have considerable impact on the use of such networks for shape-memory behaviour.
:4 diamine:diacrylate molar ratio) during AZ2 (Eg is the glassy modulus at 25 °C, Er is the rubbery modulus). tan(δ) and storage modulus curves are reported in ESI
| AZ2 progress (days) | TDMAg (°C) | Eg at 25 °C (GPa) | Er at 90 °C (MPa) | ΔTg (°C) | tan(δ) | tan(δ) at half width (°C) |
|---|---|---|---|---|---|---|
| 0 | 55.0 ± 1.0 | 1.76 ± 0.12 | 15.3 ± 0.6 | 28.0 ± 1 | 0.67 ± 0.02 | 23.0 ± 0.8 |
| 2 | 58.7 ± 0.6 | — | 16.6 ± 0.5 | 16.7 ± 0.5 | 0.66 ± 0.01 | — |
| 6 | 65.5 ± 0.7 | 1.88 ± 0.09 | 24.0 ± 1.5 | 10.2 ± 0.6 | 0.58 ± 0.01 | 18.1 ± 1 |
| 12 | 69.1 ± 0.2 | 1.90 ± 0.08 | 27.3 ± 0.4 | 9 ± 0.1 | 0.52 ± 0.01 | 16.4 ± 0.2 |
| 25 | 69.0 ± 0.4 | 1.98 ± 0.04 | 27 ± 0.5 | 9 ± 0.2 | 0.52 ± 0.01 | 15.2 ± 0.3 |
To highlight the role of AZ2 reaction in SMP, a typical shape-memory creation procedure (SMCP) was done using a cyclic thermomechanical tensile test.6 A strain-controlled procedure was designed as follow.1,6 First, the sample was heated to the deformation temperature Td = 90 °C at a rate of 3 °C min−1 and equilibrated for 7 min. Then the sample was stretched to a maximal strain value εm = 5% at a strain rate of 2.5% min−1. Later, under the imposed strain, the sample was cooled from Td to the set temperature Tc = 25 °C at a rate of 10 °C min−1 and equilibrated for 7 min. Then, deformation is released by setting the stress σ to 0 MPa at Tc.
Creep or spontaneous recovery of the permanent shape upon unloading is recorded via the deformation resulting from the fixing step defined as εu. Finally, the temporarily deformed sample is heated to Td (σ = 0 MPa). Irrecoverable deformation transmitted to the sample resulting from the recovery step is recorded via εp. This four-step thermomechanical cycle was repeated four times (N = 4, N stands for the cycle number) for each sample to ensure reproducibility.
A typical cyclic thermomechanical test (CTT) of the simultaneous monitoring of temperature, stress and strain is shown in Fig. 1 before and after AZ2 reaction. It can be noticed that full completion of AZ2 reaction leads to improved shape memory performance. Only a small residual strain is still observed upon completion of the cycle, representing the slight molecular anisotropy permanently created along the deformation axis.16 From these experiments, the shape memory behaviour was assessed by calculating the shape fixity (Rf), the shape recovery (Rr) and the recovery rate (νr) using eqn (1), (2) and (3).
| Rf(N) = εu(N)/εm | (1) |
| Rr(N) = (εu(N) − εp(N))/(εm(N) − εp(N − 1)) | (2) |
| νr = Rr/ΔTrec | (3) |
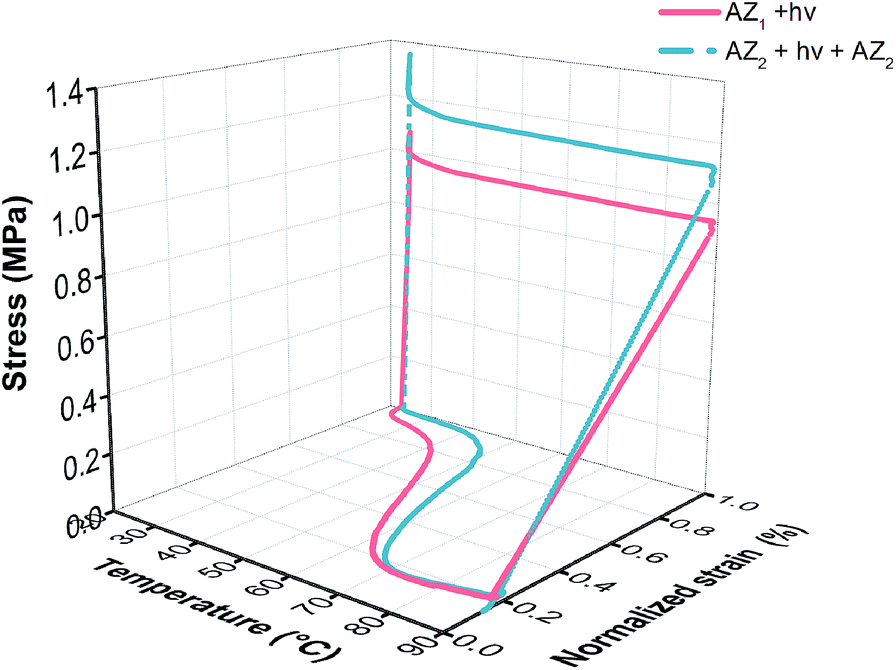 | ||
| Fig. 1 Resulting σ–T–ε curves obtained from cyclic thermomechanical test (strain controlled programing) of three-step based polymer network (1:4 amine:acrylate molar ratio) before and after AZ2. | ||
:4 amine:acrylate molar ratio) before and after AZ2
| Rf (%) | Rr (%) | νr (% °C−1) | |
|---|---|---|---|
| Before AZ2 (t = 0 d) | |||
| Cycle 1 | 88.7 | ||
| Cycle 2 | 92.4 | 89.7 | 2.5 |
| Cycle 3 | 93.9 | 91.8 | 2.8 |
| Cycle 4 | 94.8 | 93 | 3.0 |
|
|||
| After AZ2 (t = 25 d) | |||
| Cycle 1 | 95.5 | ||
| Cycle 2 | 96.0 | 95.3 | 4.0 |
| Cycle 3 | 96.0 | 94.6 | 4.0 |
| Cycle 4 | 96.1 | 94.9 | 4.0 |
All Rf, Rr and νr values were found to increase after AZ2 reaction. In particular, the shape fixity Rf grows from 88.7 to 95.5% (cycle 1). As stated in literature,20 the shape fixity of SMP is governed by two main factors: the modulus at room temperature and the fraction of mobile chains. Indeed, when the network is stretched during a CTT, orientation of stretched chains is actually preserved by vitrification of the soft segment in the cooling stage. Nevertheless, a given fraction of the chains does not vitrify and keeps their mobility. These mobile chains generate a retractive force upon removal of the tensile load due to entropic elasticity. As reported in Table 2, the glassy modulus Eg at 25 °C does not significantly evolve through AZ2. By contrast, the fraction of mobile chains decreases (higher network homogeneity) upon AZ2 crosslinking and hence increases Rf. Regarding Rr, it increased from 89.7 to 95.3% (cycle 2). This result is directly related to the increase in rubbery modulus occurring during AZ2. Indeed, a high Er provides high elastic recovery at high temperature. More significantly, an increase of the recovery rate νr from 2.5 (before AZ2) to 4.0% °C−1 (after AZ2) is observed. This trend is explained by the improvement in network homogeneity (reduction of Mc and mobile chains) which enables to drastically shorten temperature range at which the mechanical transition occurs. Thus, this synthesis strategy allows designing photo-induced SMP exhibiting stable shape fixing and a fast and complete shape recovery. Finally, it should be observed that thermal treatment after AZ1 + hν fosters AZ2 as Rf and Rr gradually increase after each cycle (Table 3). This opens up the possibility to achieve final SMP in a shorter time.
To explain the AZ2 reaction mechanism over time, the evolution of Rf and νr is plotted in Chart 1 together with acrylate conversion that was evaluated in a previous study.13 From Chart 1, it can be stated that Rf reaches its final value after 4 days. However, both νr and acrylate conversion still did not level off and continue to evolve up to 12 days, νr increasing from 3.3 to 4% °C−1 and acrylate conversion from 88 to 91%. This difference explains how AZ2 influences SMP properties by acting through two levers: (1) reducing fraction of mobile chains and (2) reducing Mc. Thus, up to 4 days, Rf and νr are improved through the joint action of both levers. After that reaction time, Rf stops to evolve meaning that mobile chains are reduced to a maximum and are no longer influent. Therefore, νr increases by means of the reduction of Mc. In conclusion, AZ2-action mechanism relies on two complementary driving forces which make photopolymers able to achieve both temporary shape fixing and permanent shape recovery within a narrow temperature range.
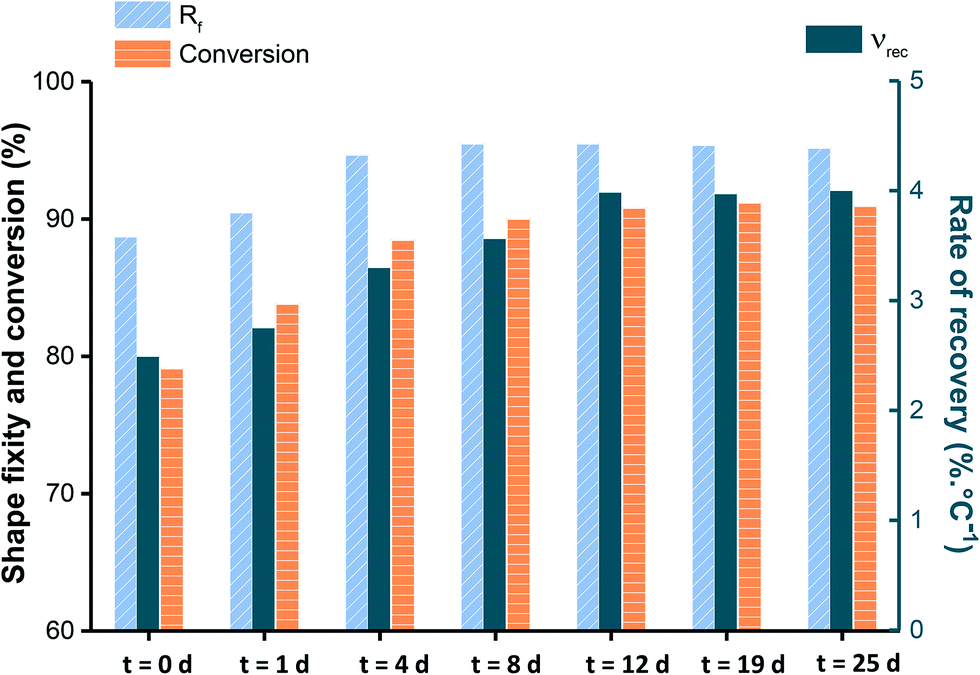 | ||
| Chart 1 Acrylate conversion, Rf and νr calculated respectively for cycle 1 and 2 at different reaction times during AZ2. | ||
Conclusions
This three-step approach highlights the interest of aza-Michael additions combined with photopolymerization to synthetize a new type of SMP. The key feature of the process is the reduction of dangling ends by AZ2 that enabled high-density cross-linking and more uniform response of the polymer chains to thermal stimuli. The range of potential monomer structures would make this process versatile and open up new horizons. SMP can be designed for a vast range of applications as thermomechanical and shape memory properties are tailorable.Notes and references
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- C. Liu, H. Qin and P. T. Mather, J. Mater. Chem., 2007, 17, 1543–1558 RSC.
- T. Xie, Polymer, 2011, 52, 4985–5000 CrossRef CAS.
- G. J. Berg, M. K. McBride, C. Wang and C. N. Bowman, Polymer, 2014, 55, 5849–5872 CrossRef CAS.
- J. Hu, Shape Memory Polymers: Fundamentals, Advances and Applications, Smithers Rapra Technology, 2014 Search PubMed.
- T. Sauter, M. Heuchel, K. Kratz and A. Lendlein, Polym. Rev., 2013, 53, 6–40 CrossRef CAS.
- C. M. Yakacki, R. Shandas, D. Safranski, A. M. Ortega, K. Sassaman and K. Gall, Adv. Funct. Mater., 2008, 18, 2428–2435 CrossRef CAS PubMed.
- W. Voit, T. Ware, R. R. Dasari, P. Smith, L. Danz, D. Simon, S. Barlow, S. R. Marder and K. Gall, Adv. Funct. Mater., 2010, 20, 162–171 CrossRef CAS.
- N. Lakhera, C. M. Yakacki, T. D. Nguyen and C. P. Frick, J. Appl. Polym. Sci., 2012, 126, 72–82 CrossRef CAS.
- R. Schwalm, in UV Coatings, Elsevier, Amsterdam, 2007, pp. 19–61 Search PubMed.
- D. P. Nair, N. B. Cramer, T. F. Scott, C. N. Bowman and R. Shandas, Polymer, 2010, 51, 4383–4389 CrossRef CAS PubMed.
- D. P. Nair, N. B. Cramer, J. C. Gaipa, M. K. McBride, E. M. Matherly, R. R. McLeod, R. Shandas and C. N. Bowman, Adv. Funct. Mater., 2012, 22, 1502–1510 CrossRef CAS.
- M. Retailleau, A. Ibrahim, C. Croutxé-Barghorn, X. Allonas, C. Ley and D. Le Nouen, ACS Macro Lett., 2015, 1327–1331 CrossRef CAS.
- A. J. Scott, A. Nabifar and A. Penlidis, Macromol. React. Eng., 2014, 8, 639–657 CrossRef CAS.
- J. E. Mark and B. Erman, Rubberlike Elasticity: A Molecular Primer, Cambridge University Press, 2007 Search PubMed.
- I. A. Rousseau and T. Xie, J. Mater. Chem., 2010, 20, 3431–3441 RSC.
- A. R. Kannurpatti, J. W. Anseth and C. N. Bowman, Polym. J., 1998, 39, 2507–2513 CAS.
- C. Arends, Polymer Toughening, Taylor & Francis, 1996 Search PubMed.
- N. Y. Choi, S. Kelch and A. Lendlein, Adv. Eng. Mater., 2006, 8, 439–445 CrossRef CAS.
- I. S. Gunes, F. Cao and S. C. Jana, Polymer, 2008, 49, 2223–2234 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods used for this study. See DOI: 10.1039/c6ra07610f |
| This journal is © The Royal Society of Chemistry 2016 |