
Density functional theory (DFT) studies of CO oxidation reaction on M13 and Au18M clusters (M = Au, Ag, Cu, Pt and Pd): the role of co-adsorbed CO molecule†
Abstract
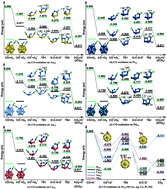
Using the icosahedra M13 (M = Au, Ag, Cu, Pt, Pd) and hetero-atom doped Au18M (M = Ag, Cu, Pt, Pd) clusters as model systems, we have systematically investigated the role of the co-adsorbed CO molecule played in the CO oxidation reaction on the basis of density functional theory (DFT) calculations. The results indicate that the co-adsorbed CO molecule at a triangular active site can induce the dissociation of the OCOO* intermediate via a tri-molecular reaction route. This mechanism is also validated on other larger single doped gold alloy clusters such as AunAg and AunCu (n = 32–34, 54). The underlying reason for promoting the oxidation effect of a co-adsorbed CO molecule is unraveled. It is found that the relatively weaker d–π* back bonding of CO on group 11 elements like Au, Ag and Cu may increase its electrophilic activity, which can facilitate the dissociation of nearby OCOO* intermediates. For the CO molecule that is bounded to the Pd and Pt atoms, it can also induce the dissociation of OCOO* intermediate, but shows weaker electrophilic activity. By explicitly considering the elementary reaction steps in a Kinetic Monte Carlo (KMC) simulation, we have shown that the tri-molecular reaction route is an alternative reaction channel of CO oxidation, which is competitive to the conventional bi-molecular route on a doped Au18M cluster.
 Please wait while we load your content...
Please wait while we load your content...

