
Sequential alcohol oxidation/putative homo Claisen–Tishchenko-type reaction to give esters: a key process in accessing novel biologically active lactone macrocycles†
Hugo Viana‡
a,
Elisabete P. Carreiro‡a,
Albertino Gotha,
Patrícia Bacalhauac,
Ana Teresa Caldeiraac,
Maria do Rosário Martinsac and
Anthony Joseph Burke*ab
aCentro de Química de Évora, Institute for Research and Advanced Studies (IIFA), University of Évora, Rua Romão Ramalho, 59, 7000 Évora, Portugal. E-mail: ajb@uevora.pt
bDepartment of Chemistry, School of Science and Technology, University of Évora, Rua Romão Ramalho 59, 7000-671 Évora, Portugal
cLaboratório HERCULES, Institute for Research and Advanced Studies (IIFA), Universidade de Évora, Palácio do Vimioso, Largo do Marquês de Marialva, 8, 7000-809 Évora, Portugal
First published on 27th June 2016
Abstract
We report an efficient methodology for the direct oxidative esterification of primary alcohols to diether-esters using pyridinium chlorochromate (PCC). Numerous studies were carried out to probe the reaction mechanism and at the same time optimize the reaction conditions. The reaction could be conducted with 1 equivalent of PCC and 1 equivalent of BF3·OEt2. Indications based on literature precedent were that the reaction may proceed via a sequential alcohol oxidation to the aldehyde followed by a putative Cr or boron catalyzed Claisen–Tishchenko-type reaction. Using this efficient methodology, we synthesized a family of novel diether-esters in very good yields; some of these molecules were subsequently tested against both acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). In addition, we also disclose a new synthetic strategy for the synthesis of lactam macrocycles with potential biological activity. This methodology included the regioselective borylation of the ester substrate and a subsequent Suzuki–Miyaura coupling to obtain the desired lactam macrocycle.
1. Introduction
Chemists have been involved with the synthesis of esters for more than 100 years.1 Classical esterification methods rely on alcohol acylation reactions with carboxylic acids and derivatives.2 Typically, carboxylic halides or anhydrides are used as the acylating agent, and thus need to be synthesized first making them less economical processes.3For this reason there have been constant efforts at developing other methods. One method is via oxidative esterification of aldehydes employing transition metals as catalysts. MnO2 has been used as the oxidizing reagent, where several aliphatic aldehydes couple with specific alcohols to form esters in good yields, despite, the substrates being limited to saturated aldehydes.4a Methyltrioxorhenium(VII) has also been used in conjunction with hydrogen peroxide (H2O2) as the oxidant. A co-catalyst such as bromide or chloride ions was additionally required.4b Patel and Gopinath reported important improvements to this procedure, with the employment of vanadium pentoxide as the catalyst and hydrogen peroxide as the oxidant.4c Alternatively, there are also some metal free procedures available. Scheidt and Maki reported that carbenes could be employed in the catalytic oxidative transformation of aldehydes to esters.4d De Luca and co-workers reported the use of trichloroisocyanuric acid for the metal-free oxidation of aldehydes to esters.4e The Tishchenko reaction (also known as the Claisen–Tishchenko reaction) leads to the conversion of aldehydes to dimeric monofunctional esters in the presence of variety of Lewis acid catalysts which include metal alcoholates like, aluminium alcoholates and magnesium alcoholates (Scheme 1).5 In the generally accepted mechanism a hydride shift takes place from one aldehyde to the other forming a metal alcoholate and an activated acyl electrophile, which couple to form the ester adduct. It is a redox process5 (note boron containing Lewis acids, like boric acid can be used5f). The reaction is atom-economical. A hydride version was reported by Werner and Koch.5h
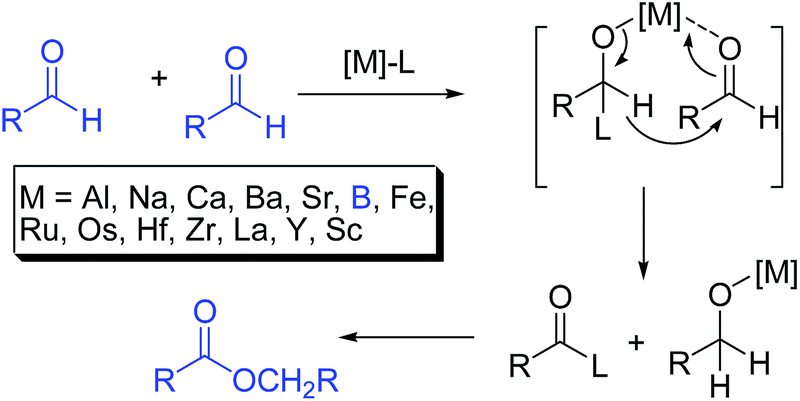 | ||
| Scheme 1 The homo Claisen–Tishchenko reaction and its generally accepted mechanism.5 | ||
Alternatively, the aldehyde can be formed in situ from the corresponding primary alcohol. Iridium is commonly known for its ability to catalyse hydrogen transfer reactions. In 2005, Katoh and co-workers reported the use of an iridium catalyst for the selective oxidation of alcohols. In this procedure, 2-butanone was used as the oxidant.6a Lei and co-workers used PdCl2(PPh3)2 for the transformation of alcohols to esters, where benzyl chloride was used as the oxidant and used to generate a key benzyl ligand for palladium, but the reaction had to be heated.6b In 2011, the first palladium-catalysed direct aerobic oxidative esterification of benzylic alcohols with methanol and several long-chain aliphatic alcohols was reported independently by Beller's6c and Lei's groups.6d Similar work on the so-called metal-catalysed oxidative cross-esterification between two alcohols was reported by Maiti and Lahiri's groups, using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and tetra-n-butylammonium bromide with oxone and Fe(OAc)2/2,6-pyridinedicarboxylic acid.6e Gao, Lei and co-workers reported similar work using PdCl2(PPh3)2 and benzyl chloride as the oxidant.6f
In 1976, Corey and Suggs reported the application of pyridinium chlorochromate (PCC, and also known as Corey's reagent) for the selective oxidation of primary alcohols,7 and since this date this has proved to be a standard method for this type of transformation. However, prior to the discovery of this transformation, Craig and Horning reported the direct oxidation of primary alcohols to esters using a chromic acid–sulphuric acid solution.8 The putative reaction pathway was considered to be via oxidation of a hemi-acetal intermediate. In 1975, Fraser-Reid and coworkers reported an oxidative esterification of some carbohydrate substrates bearing a hydroxymethyl group using Collins reagent – CrO3·2C5H5N.9 The reaction afforded dimeric ester products, which was considered anomalous at the time. These authors considered a Tishchenko-type reaction mechanism to be at work. In 2001, Potier and coworkers reported the formation of an ester derived from both (S)-2,3-O-isopropylideneglyceraldehyde and un-oxidised (S)-2,3-O-isopropylideneglycerol, it was suggested that the putative hemiacetal intermediate was rapidly oxidized to the ester product.10 On the basis of this report by Potier's group, in 2003 Fraser-Reid and co-workers substantiated their claim of formation of the dimeric ester via a Tishchenko type mechanism.5,11
In 2005, Hunsen reported the formation of phenethyl 2-phenylacetate, upon oxidizing 2-phenylethanol to its corresponding aldehyde derivative, using a mixture of PCC and periodic acid as oxidant and co-oxidant.12
In a similar vein, to some of these latter reports in this paper we report our results on an efficient simple process that provides dimeric esters from primary alcohols using PCC as the oxidant. Furthermore, these esters can be easily harnessed to afford macrocycles with potential application in medicinal chemistry and in drug discovery (see below).11 Known macrocyclic drugs are almost exclusively derived from natural sources (mostly from microorganisms) and are either identical to or closely related to naturally occurring macrocycles.
2. Results and discussion
In a study directed at the preparation of boronic-ester aldehydes for novel metal-catalysed cyclizations, we encountered much difficulty at oxidizing linear non-bulky aryl alkyl alcohols to the corresponding aldehydes using PCC. Instead, the alcohol substrate underwent an oxidative esterification to provide the corresponding symmetric ester, in a similar fashion to the observations of Hunsen,12 Fraser-Reid,9,11 Potier10 and Craig.8 For that, we observed that when 2-(2-bromophenoxy)ethanol (1a) was reacted with PCC it gave (2a) as the major product in 61% yield (in two steps) and not the expected aldehyde which was obtained in only low quantities (Scheme 2). 2′-Bromophenoxyacetaldehyde was isolated from the silica gel column in low yield (<4%). Celite was added to the reaction mixture as it facilitates greatly the reaction work-up, by immobilizing the unwanted toxic Cr salts. | ||
| Scheme 2 Oxidative esterification of (1a) to (2a) using PCC. | ||
In order to study the scope of this reaction, a number of alcohols had to be synthesized. These ether alcohols (1a–j) were synthesized by etherification of the corresponding phenol precursors with bromoethanol (Scheme 3). They were obtained in yields in the range 33% to 95%. Of note was the selective synthesis of compound (2f) from its pyridinol substrate, gratifyingly and very interesting as the product of possible substitution in the 2-position by the bromoalcohol reagent was not obtained.
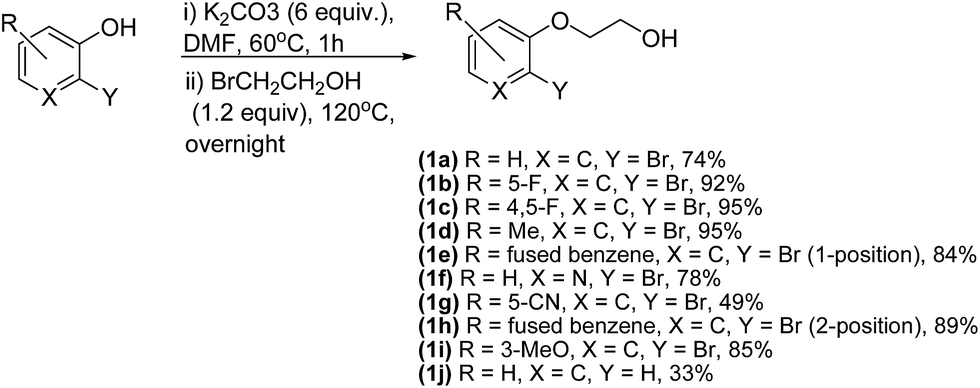 | ||
| Scheme 3 Etherification of phenol derivatives (1a–j). | ||
2.1. Preliminary studies
In order to understand the specific mechanism of this reaction we initiated a series of studies that involved screening and optimizing the reaction conditions. We looked at: (1) the PCC loading, (2) the reaction solvent and (3) the type of alcohol substrate. Table 1 shows the results of the PCC loading experiments.The results show that the minimum amount of PCC required for high conversion is 2 equivalents (Table 1, entry 2). If less than this quantity is used (1 equivalent), the conversion of the reaction decreases significantly (Table 1, entry 3, 48% with 1 equivalent). With less than 1 equivalent the conversion was very poor or no reaction was observed (Table 1, entries 4 and 5). On the other hand, if more than 2 equivalents are used (4 equivalents), the conversion of the reaction is not significantly affected (Table 1, entry 1).
In addition, we conducted a solvent screening study. Different types of solvents, including apolar protic solvents (DMF, MeCN), mild polar aprotic solvents (DCM, THF and dioxane) and the apolar solvent toluene were tested under the conditions shown in Table 2.

This study indicated that only DCM (dichloromethane) was effective in this reaction (Table 2, entry 6). Both DMF and dioxane also supported the reaction, but to a lesser extent than DCM. When toluene and MeCN were employed (Table 2, entries 3 and 4) there was no reaction and we only detected starting material in the 1H NMR spectrum. In the case of THF, a moderate amount of aldehyde intermediate was detected (Table 2, entry 5). In conformity with the literature for PCC oxidations, DCM is the preferred solvent.7 One possible explanation might be the optimal solubilizing properties of this solvent, promoting maximum diffusion and mass transfer of the main reactants during the course of the reaction.
2.2. 1H NMR reaction kinetic study
We also examined the kinetic profile of this reaction (oxidative esterification of (1a) to (2a)). This was accomplished by performing a 1H NMR kinetic experiment. We prepared our samples as follows: 0.5 mL of deuterated chloroform were added to an NMR tube under nitrogen. After this, the alcohol and PCC were also added to the tube. The 1H NMR spectra were taken on an hourly basis for 20 hours. Fig. 1 shows the formation of product as a function of time. Analysis of this data showed an almost linear relationship for the formation of the product over time. | ||
| Fig. 1 Reaction kinetic studies for the transformation of (1a) into (2a). | ||
These results show that this reaction formed product at an almost constant rate during the entire experiment.
The 1H NMR spectra for the reaction at the beginning and at the end with this substrate (1a) are shown in Fig. S34 (ESI†).
In order to probe further the reaction mechanism, we treated 2-(2-bromophenoxy)ethanol (1a) with 3-bromopropan-1-ol in the presence of 2 equivalents of PCC. Unfortunately, although the substrate was totally consumed, the reaction failed to provide the desired product. We also reacted (1a) with methanol with the hope of forming the methyl ester, but the substrate failed to react.
2.3. Proposed mechanism and complementary studies
The mechanism of this reaction is intriguing and has been the subject of literature discussion over the last 10 years or so. There are two possibilities. The first involves partial formation of the aldehyde that is attacked by the alcohol substrate to form a hemi-acetal intermediate.10,11 As the conditions are slightly acidic this should be acid catalysed and in fact catalysed by BF3 when the reaction is run in the presence of this Lewis acid. The second part may involve oxidation of the hemi-acetal to the ester as proposed by Potier's group.10 This might be achieved by formation of a chromium alcoholate derivative of the hemi-acetal that undergoes β-elimination do form the ester carbonyl with expulsion of a reduced chromium species. The second possibility (Scheme 3) involves the standard Claisen–Tishchenko reaction as alluded to by Fraser-Reid and co-workers.9,11 In this case we suspect that after formation of the aldehyde in situ a Cr coordinated hemi-acetal is formed, which then tethers via a Cr bridge with an aldehyde molecule, this complex then undergoes hydride transfer to give the ester product molecule and a putative Cr-alcoholate species that can then attack the aldehyde intermediate to start another catalytic cycle. Chromium alcoholate species are known.13 In fact, we conducted the reaction with 1 equivalent of trifluoroboron etherate and 1 equivalent of PCC and got very good results (see discussion below and Table 3) (this was also a strong indication of a Tishchenko type reaction mechanism).
|
||||
|---|---|---|---|---|
| Entry | Substrate | X, Y, R | Product | Yield/% |
| a Determined by 1H NMR with the addition of an internal standard (mesitylene).b Using 1 equiv. of PCC and 1 equiv. of BF3·OEt2. | ||||
| 1 | (1a) | X = C, Y = Br | (2a) | 61a |
| 2b | (1a) | X = C, Y = Br | (2a) | 61 |
| 3 | (1b) | X = C; R = 5-fluor, Y = Br | (2b) | 76a |
| 4 | (1c) | X = C; R = 4,5-difluor, Y = Br | (2c) | 72a |
| 5 | (1d) | X = C; R = 4-methyl, Y = Br | (2d) | 47 |
| 6 | (1e) | 1-Bromonaphthalene | (2e) | 75 |
| 7 | (1f) | X = N, Y = Br | (2f) | 56 |
| 8 | (1g) | X = C; R = 4-nitrile, Y = Br | (2g) | 59 |
| 9 | (1h) | 2-Bromonaphthalene | (2h) | 73 |
| 10 | (1i) | X = C; R = 3-methoxy, Y = Br | (2i) | 46 |
| 11 | (1j) | X = C, Y = H | (2j) | 65 |

In order to probe the reaction mechanism, we conducted some key experiments. As can be seen from Scheme 4, outlining one of the possible mechanisms of this reaction, we suggest hydride transfer from the Cr-alcoholate to the aldehyde. In order to investigate this step and thus support our mechanistic postulate, we carried out the reaction with (1e) (which reacts well in this reaction, see entry 6, Table 3) under the conditions described previously, but with 2 equivalents of 2-chlorobenzaldehyde as a sacrificial oxidant. Our reasoning was that the sacrificial aldehyde would compete with 2′-bromophenoxyacetaldehyde for the hydride liberated by the Cr-alcoholate to give 2-chlorophenylmethanol, the presence of the latter would support this step. Unfortunately, we failed to detect any 2-chlorophenylmethanol in the reaction mixture, obtaining only the ester product (2e) in the same yield as obtained previously. As this was an aromatic aldehyde (although activated) it was probably outcompeted by the 2′-bromophenoxyacetaldehyde, which is a less sterically hindered aldehyde.
 | ||
| Scheme 4 Proposed mechanism for the putative Cr-catalysed Claisen–Tishchenko reaction. | ||
Supported by the 1H NMR kinetic experiments, we clearly observed that the amount of aldehyde species remains constant in the mixture during the entire reaction, indicating slow oxidation of the starting alcohol, and this reaction step probably competes with the Cr-catalysed Claisen–Tishchenko reaction.
In order to probe the mechanism further, we also carried out a series of reactions using the conditions described in Scheme 2, between (2a) and a series of alcohols like, benzyl alcohol, isopropanol, 3-bromopropanol, but we obtained a very complex mixture of products, indicating poor reaction selectivity thus precluding this strategy for the synthesis of the desired ester products.
2.4. Evaluating the reaction scope
After having established the best conditions for carrying out this oxidative/Cr-catalysed putative Claisen–Tishchenko reaction, we embarked on studying the reaction scope using a set of different alcohol substrates. Using the conditions described in Scheme 2, 8 additional diether-esters ((2c)–(2j)) were synthesized (Table 3).The overall yields of these reactions from two steps were very good, and although column chromatographic purifications where required to isolate the products, these reactions were generally quite clean, but they always contained small amounts of the aldehyde intermediate. All these compounds were obtained as light coloured solids. Comparing the reactivity of these compounds, we observed that molecules bearing electron-withdrawing groups (with the exception of the nitrile, Table 3, entry 8) in the benzene ring and specifically the disubstituted example (Table 3, entry 4) gave the best results. The same was observed for those compounds containing a naphthyl ring (Table 3, entry 6 and 9). However, in the case of molecules bearing electron-donating groups (Table 3, entry 5 and 10) the yields were lower. When the substrate containing an electron withdrawing pyridine ring was used a yield of 59% was obtained (Table 3, entry 7). The non-bromine substituted substrate (1j) was evaluated and it afforded a good yield of 65% (Table 3, entry 11) equivalent to the results obtained using the electron-withdrawing group containing substrates.
With strong indications that there was a Claisen–Tishchenko reaction at work here, we reacted compounds (1a) with 1 equivalent of PCC and 1 equivalent of the Lewis acid (BF3·OEt2) under the conditions shown in Scheme 2. The diether-ester (2a) were obtained in good yield (contrary to the 48% conversion obtained previously using just 1 equivalent of PCC) (Table 1, entry 3).
We were also interested in developing a crossed-variant of this reaction to improve reaction scope and product diversity. We treated the aldehyde intermediate 2-bromophenoxyacetaldehyde (Scheme 2) with a number of different commercially available alcohols (like, benzyl alcohol, allylic alcohol, 2-bromopropanol and iso-propanol) under the same reaction conditions described in Scheme 2. Unfortunately, after purification and 1H NMR analysis of the reaction mixtures, we observed no or very small amounts of the initial aldehyde substrate in the reaction mixture and no signs of the ester products either. Selectivity issues were probably at play here, perhaps the different alcohols that were tested in these studies underwent oxidation at a significantly faster rate than (1a) to give aldehydes with no possibility for the Claisen–Tishchenko reaction step.
We have also shown that these ester products can be selectively borylated and cyclized to give macrocyclic compounds. Macrocyclic compounds are currently of interest due to their medicinal properties.14 Compound (2a) was borylated to give compound (3) (Scheme 5) in very good yield, this was then cyclized via a Suzuki–Miyaura coupling to give the macrocyclic lactone (4).15
 | ||
| Scheme 5 Synthesis of macrocyclic lactone (4) via a borylation/Suzuki–Miyaura coupling sequence from diether-ester (2a).15 | ||
2.5. Biological assays
Since our group has a strong interest in the design and synthesis of cholinesterase inhibitors for targeting Alzheimer's disease, and considering the structural aspects of compounds (2) and the macrocycle (4) – bearing both key aromatic units for π–π interactions with active site aromatic amino acid residues and the ester group to H-bond with the H-bonding catalytic triad residues14 – we decided to evaluate their inhibition against the standard model cholinesterases (ChEs); eel acetylcholinesterase (EeAChE) and equine serum butyrylcholinesterase (EqBuChE) (Table 4). The dual inhibitor rivastigmine was used as the standard.| Entry | Compound | IC50 AChEa (μM) | IC50 BuChEa (μM) | ||
|---|---|---|---|---|---|
| Without incub. | With incub. (60 min) | Without incub. | With incub. (15 min) | ||
| a IC50 values are expressed as mean ± SD (n = 3) based on dose–response curves, using the Origin 8.0 Pro. | |||||
| 1 | (2b) | 123.24 ± 2.57 | 67.95 ± 8.21 | 863.12 ± 125.50 | 664.10 ± 11.76 |
| 2 | (2c) | 150.51 ± 4.29 | 107.25 ± 0.69 | 951.31 ± 129.02 | 481.79 ± 20.09 |
| 3 | (2e) | 88.84 ± 7.84 | 38.37 ± 1.38 | >1500 | 437.85 ± 10.15 |
| 4 | (4) | 85.12 ± 5.12 | 109.95 ± 13.54 | 336.80 ± 55.83 | 423.45 ± 145.54 |
| Standard | Rivastigmine | 342.50 ± 13.00 | 127.60 ± 5.90 | 26.71 ± 3.77 | 1.38 ± 0.01 |
All the tested compounds exhibited better inhibitions for AChE than for BuChE, thus being more selective for this enzyme. The incubation time was also studied with both enzymes, and was found to be crucial to the inhibition process, except for compound (4) that showed in fact poorer binding to the enzyme active site, as attested to by the increase in the IC50 values (Table 4, entry 4).
In the case of AChE, all compounds presented better inhibition than the standard, rivastigmine. Compound (2e) provided the best IC50 value of 38.37 μM, showing the importance not only of the aromatic rings for binding via π–π interactions, but also the ester group that could be hydrolysed by the catalytic triad in the enzymes' active site.14
In the case of BuChE, compound (4) gave the best IC50 value of 336.80 μM, followed by (2e), showing again the importance of π–π interaction of the aromatic rings. Overall these compounds are much better inhibitors for AChE than for BuChE which makes sense when one considers the tighter active site that exists in the case of the former.16
3. Conclusions
In this paper, we reported the application of a sequential PCC alcohol oxidation/putative Cr-catalysed Claisen–Tishchenko reaction to provide interesting diether-esters. Numerous studies were carried out to probe the reaction mechanism and at the same time optimize the reaction conditions. These included oxidant loading, solvent screening and kinetic studies. It was established that 1 equivalent of PCC and 1 equivalent of BF3·OEt2 were the best combination of reagents to be employed. Using this methodology, a family of novel diether-esters was synthesized in very good yields; one was then selectively borylated and after Suzuki–Miyaura coupling transformed into a macrocyclic lactone. Some of these molecules including the macrocycle were subsequently screened for both AChE and BuChE inhibition, and showed potential for AChE inhibition. Unfortunately, we were unable to provide substantial support for our mechanistic proposal involving a purported Cr-catalysed Claisen–Tishchenko reaction, despite our best efforts. We are currently investigating a catalytic version of this reaction and other less toxic replacements of PCC.4. Experimental
4.1. Materials and methods
All reagents were obtained from Aldrich, Acros and Alfa Aesar and were used as obtained. When appropriate, the solvents used were dried using current laboratory techniques. In addition, all reactions were conducted under a nitrogen atmosphere. Column chromatography purifications was carried out on silica gel (sds, 70–200 μm). Thin layer chromatography (TLC) was carried out on aluminium backed Kiselgel 60 F254 plates (Merck), which were visualized either by UV light or revealed by phosphomolybdic acid in ethanol. The NMR analyses were recorded on a Bruker Avance III instrument (400 MHz) using CDCl3 as solvent and the signal from the residual CHCl3 as an internal standard. Mass spectra were obtained from C.A.C.T.I., at Universidade de Vigo, on a Waters-Micromass (MicroTOF, ESI) or FAB Focus (Bruker Daltonics), using the TOF technique and at the University of Salamanca where a Waters ZQ4000 quadrupole mass spectrometer was used for ESI analysis. HMRS analysis were obtained from the University of Potsdam, Germany, on a ESI-Q-time-of-flight (TOF) mass analyzer (Micromass/Waters, Manchester, UK).The modified Ellman method was used for the bioassay studies (see the ESI†).17
4.2. Synthetic procedures
2-(2-Bromophenoxy)ethanol (1a). Following the general procedure, 2-bromophenol (0.38 g, 2.18 mmol), K2CO3 (1.81 g, 13.0 mmol) and 2-bromoethanol (0.19 mL, 2.62 mmol) were dissolved in DMF (10 mL) in a round bottom flask and allowed react as described above. After purification by silica gel chromatography (Hex
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc (2:1)) compound (1a) was obtained as a pale yellow oil (0.37 g, 74%). 1H NMR (400 MHz, CDCl3) δ: 2.16 (sbroad, 1H, OH), 3.99 (t, J = 4.6 Hz, 2H, CH2), 4.15 (t, J = 4.5 Hz, 2H, CH2), 6.87 (t, J = 7.7 Hz, 1H, ArH), 6.92 (d, J = 8 Hz, 1H, ArH), 7.25–7.29 (m, 1H, ArH), 7.54 (d, J = 7.8 Hz, 1H, ArH).18
:EtOAc (2:1)) compound (1a) was obtained as a pale yellow oil (0.37 g, 74%). 1H NMR (400 MHz, CDCl3) δ: 2.16 (sbroad, 1H, OH), 3.99 (t, J = 4.6 Hz, 2H, CH2), 4.15 (t, J = 4.5 Hz, 2H, CH2), 6.87 (t, J = 7.7 Hz, 1H, ArH), 6.92 (d, J = 8 Hz, 1H, ArH), 7.25–7.29 (m, 1H, ArH), 7.54 (d, J = 7.8 Hz, 1H, ArH).18
2-(2-Bromo-5-flourophenoxy)ethanol (1b). Following the general procedure, 2-bromo-5-flourophenol (0.6 g, 3.14 mmol), K2CO3 (2.6 g, 18.8 mmol) and 2-bromoethanol (0.296 mL, 3.77 mmol) were dissolved in DMF (10 mL) in a round bottom flask and allowed react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (1b) was obtained as a colorless oil (0.607 g, 92%). 1H NMR (400 MHz, CDCl3) δ: 2.09 (sbroad, 1H, OH), 4.01 (t, J = 4.5 Hz, 2H, CH2), 4.12 (t, J = 4.5 Hz, 2H, CH2), 6.60–6.68 (m, 2H, ArH), 7.46–7.50 (m, 1H, ArH).19
2-(2-Bromo-4,5-diflourophenoxy)ethanol (1c). Following the general procedure, 2-bromo-4,5-flourophenol (0.66 g, 3.14 mmol), K2CO3 (2.6 g, 18.8 mmol) and 2-bromoethanol (0.296 mL, 3.77 mmol) were dissolved in DMF (10 mL) in a round bottom flask, and allowed react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (1c) was obtained as a colorless oil (0.75 g, 95%). 1H NMR (400 MHz, CDCl3) δ: 2.18 (sbroad, 1H, OH), 3.99 (t, J = 4 Hz, 2H, CH2), 4.08 (t, J = 4 Hz, 2H, CH2), 6.79 (dd, J = 12, 7 Hz, 1H, ArH), 7.39 (t, J = 9 Hz, 1H, ArH). The NMR data is coherent with the literature.20
2-(2-Bromo-4-methylphenoxy)ethanol (1d). Following the general procedure, 2-bromo-4-methylphenol (0.6 g, 3.21 mmol), K2CO3 (2.66 g, 19.2 mmol) and 2-bromoethanol (0.28 mL, 4.04 mmol) were dissolved in DMF (10 mL) in a round bottom flask and allowed react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (1d) was obtained as a pale yellow oil (0.706 g, 95%). 1H NMR (400 MHz, CDCl3) δ: 2.28 (sbroad, 1H, OH), 3.97 (t, J = 4 Hz, 2H, CH2), 4.11 (t, J = 4.5 Hz, 2H, CH2), 6.81 (d, J = 8 Hz, 1H, ArH), 7.05 (d, J = 8 Hz, 1H, ArH), 7.37 (s, 1H, ArH).21
2-(1-Bromonaphth-2-yloxy)ethanol (1e). Following the general procedure, 2-bromonaphthanol (0.5 g, 2.24 mmol), K2CO3 (1.86 g, 13.4 mmol) and 2-bromoethanol (0.198 mL, 2.83 mmol) were dissolved in DMF (10 mL) in a round bottom flask and allowed react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (1e) was obtained as a colorless solid (0.50 g, 84%). 1H NMR (400 MHz, CDCl3) δ: 2.39 (sbroad, 1H, OH), 4.03 (t, J = 4 Hz, 2H, CH2), 4.30 (t, J = 4.5 Hz, 2H, CH2), 7.27 (d, J = 8 Hz, 1H, ArH), 7.43 (d, J = 8 Hz, 1H, ArH), 7.59 (t, J = 8 Hz, 1H, ArH), 7.80 (t, J = 7 Hz, 1H, ArH), 8.22 (d, J = 9 Hz, 1H, ArH).22
2-((2-Bromopyridin-3-yl)oxy)ethanol (1f). Following the general procedure, 2-bromopyridin-3-ol (0.5 g, 2.9 mmol), K2CO3 (2.39 g, 17.4 mmol) and 2-bromoethanol (0.25 mL, 3.5 mmol) were dissolved in DMF (10 mL) in a round bottom flask and allowed react as described above. After purification by silica gel chromatography (Hex
:EtOAc (1:1)) compound (1f) was obtained as a light yellow oil (0.492 g, 78%). 1H NMR (400 MHz, CDCl3) δ: 2.16 (sbroad, 1H, OH), 4.03 (t, J = 4.5 Hz, 2H, CH2), 4.16 (t, J = 4.6 Hz, 2H, CH2), 7.17–7.24 (m, 2H, ArH), 8.01 (s, 1H, ArH).23
3-Bromo-4-(2-hydroxyethoxy)benzonitrile (1g). Following the general procedure, 3-bromo-4-hydroxybenzonitrile (0.5 g, 2.53 mmol), K2CO3 (2.1 g, 15.2 mmol) and 2-bromoethanol (0.23 mL, 3.04 mmol) were dissolved in DMF (10 mL) in a round bottom flask and allowed react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (1g) was obtained as a white solid (0.30 g, 49%). 1H NMR (400 MHz, CDCl3) δ: 2.14 (sbroad, 1H, OH), 4.04 (sbroad, 2H, CH2), 4.20 (t, J = 4.5 Hz, 2H, CH2), 6.96 (d, J = 9 Hz, 1H, ArH), 7.59 (dd, J = 9, 2 Hz, 1H, ArH), 7.84 (d, J = 2 Hz, 1H, ArH).24
2-((3-Bromonaphthalen-2-yl)oxy)ethanol (1h). Following the general procedure, 3-bromonaphthalen-2-ol (0.25 g, 1.10 mmol), K2CO3 (0.91 g, 6.60 mmol), 2-bromoethanol (0.18 g, 1.42 mmol) were dissolved in DMF (2.5 mL) in a round bottom flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (5:1)) compound (1h) was obtained as a light brown solid (0.26 g, 89%). 1H NMR (400 MHz, CDCl3) δ: 2.48 (sbroad, 1H, OH), 4.02 (t, J = 4.5 Hz, 2H, CH2), 4.28 (t, J = 4.6 Hz, 2H, CH2), 7.25 (d, J = 9 Hz, 1H, ArH), 7.43 (d, J = 8 Hz, 1H, ArH), 7.58 (t, J = 8 Hz, 1H, ArH), 7.81 (d, J = 8 Hz, 1H, ArH), 8.22 (d, J = 9 Hz, 1H, ArH). 13C NMR (100 MHz, CDCl3) δ: 61.6 (CH2), 72.1 (CH2), 110.3 (CBr), 115.8 (CH), 124.8 (CH), 126.4 (CH), 127.9 (CH), 128.1 (CH), 129.2 (CH), 130.3 (C), 133.1 (C), 153.0 (C). ESI MS (m/z): 268.80 (M + 1).
2-(2-Bromo-3-methoxyphenoxy)ethanol (1i). Following the general procedure, 2-bromo-3-methoxyphenol (0.5 g, 2.46 mmol), K2CO3 (2.04 g, 14.8 mmol) and 2-bromoethanol (0.21 mL, 2.96 mmol) were dissolved in DMF (5 mL) and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (1i) was obtained as a white oil (0.512 g, 85%). 1H NMR (400 MHz, CDCl3) δ: 2.49 (sbroad, 1H, OH), 3.87 (s, 3H, OMe), 3.96 (t, J = 4.6 Hz, 2H, CH2), 4.11 (t, J = 4.5 Hz, 2H, CH2), 6.56 (dd, J = 9, 5 Hz, 1H, ArH), 7.19 (t, J = 8 Hz, 1H, ArH). 13C NMR (100 MHz, CDCl3) δ: 56.5 (OMe), 61.3 (CH2), 71.0 (CH2), 101.8 (CBr), 105.2 (CH), 106.5 (CH), 128.4 (CH), 156.2 (C), 157.2 (C). ESI MS (m/z): 248.80 (M + 1).
2-Phenoxyethan-1-ol (1j). Following the general procedure, phenol (0.5 g, 5.3 mmol), K2CO3 (4.56 g, 31.8 mmol) and 2-bromoethanol (0.45 mL, 6.4 mmol) were dissolved in DMF (10 mL) and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (1j) was obtained as a colorless oil (0.243 g, 33%). 1H NMR (400 MHz, CDCl3) δ: 1.95 (sbroad, 1H, OH), 3.97 (t, J = 4.5 Hz, 2H, CH2), 4.09 (t, J = 4.5 Hz, 2H, CH2), 6.92–6.99 (m, 3H, ArH), 7.30 (t, J = 8 Hz, 2H, ArH).25
:1) was added to the crude product. The resulting residue was filtered over a silica pad under vacuum. The solvent was evaporated under reduced pressure. After purification by silica gel chromatography, products ((2a)–(2j)) were obtained in good to very good yields.
2-(2-Bromophenoxy)ethyl-2-(2-bromophenoxy)acetate (2a). Following the general procedure, compound (1a) (0.18 g, 0.83 mmol), celite (0.72 g) and PCC (0.36 g, 1.66 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (5:1)) compound (2a) was obtained as a white solid (0.11 g, 61%). 1H NMR (400 MHz, CDCl3) δ: 4.27 (t, J = 4 Hz, 2H, CH2), 4.64 (dd, J = 8, 4 Hz, 2H, CH2), 4.79 (s, 2H, CH2COOR), 6.82–6.94 (m, 4H, ArH), 7.15–7.23 (m, 1H, ArH), 7.28 (qd, J = 7.1, 1.6 Hz, 1H, ArH), 7.57 (ddd, J = 4.8, 3.5, 1.7 Hz, 2H, ArH). 13C NMR (101 MHz, CDCl3) δ: 63.2 (CH2), 66.2 (CH2), 67.0 (CH2), 112.5 (CBr), 112.6 (CBr), 113.9 (CH), 113.9 (CH), 122.7 (CH), 123.1 (CH), 128.5 (CH), 128.5 (CH), 133.6 (CH), 133.7 (CH), 154.4 (C), 154.8 (C), 168.4 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).ESI-TOF MS (m/z): 428.93 (M + 1). HMRS: (ESI-TOF) calcd for C16H15O4Br2 428.9313 [M + H]+, found 428.9337.
2-(2-Bromo-5-fluorophenoxy)ethyl 2-(2-bromo-5-fluorophenoxy)acetate (2b). Following the general procedure, compound (1b) (0.30 g, 1.28 mmol), celite (1.1 g) and PCC (0.55 g, 2.56 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (5:1)) compound (2b) was obtained as a white solid (0.226 g, 76%). 1H NMR (400 MHz, CDCl3) δ: 4.24 (dd, J = 8, 4 Hz, 2H, CH2), 4.67 (dd, J = 8, 4 Hz, 2H, CH2), 4.77 (s, 2H, CH2COOR), 6.63 (ddd, J = 15.3, 8.0, 4.8 Hz, 4H, ArH), 7.50 (dd, J = 8.7, 6.4 Hz, 2H, ArH). 13C NMR (101 MHz, CDCl3) δ: 63.1 (CH2), 66.1 (CH2), 67.0 (CH2), 102.0 (dd, J = 26.7, 22.8 Hz, CH × 2), 106.7 (d, J = 1.6 Hz, CO), 106.7 (d, J = 1.6 Hz, CO), 109.6 (dd, J = 57.5, 22.4 Hz, CH × 2), 133.9 (dd, J = 18.5, 9.5 Hz, CH × 2), 155.2 (d, J = 10.0 Hz, CBr), 155.6 (d, J = 10.1 Hz, CBr), 161.3 (d, J = 13.7 Hz, CF), 163.7 (d, J = 13.4 Hz, CF), 167.7 (CO).ESI-TOF MS (m/z): 464.91 (M + 1).
2-(2-Bromo-4,5-difluorophenoxy)ethyl 2-(2-bromo-4,5-difluorophenoxy)acetate (2c). Following the general procedure, compound (1c) (0.30 g, 1.19 mmol), celite (1.0 g) and PCC (0.51 g, 2.38 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (5:1)) compound (2c) was obtained as a white solid (0.217 g, 73%). 1H NMR (400 MHz, CDCl3) δ: 4.22 (t, J = 4 Hz, 2H, CH2), 4.64 (t, J = 4 Hz, 2H, CH2), 4.74 (s, 2H, CH2COOR), 6.77 (ddd, J = 11.2, 6.8, 0.9 Hz, 2H, ArH), 7.42 (ddd, J = 9.4, 8.4, 2.7 Hz, 2H, ArH). 13C NMR (101 MHz, CDCl3) δ: 63.1 (CH2), 66.9 (CH2), 67.8 (CH2), 103.5 (d, J = 21.6 Hz, CH × 2), 104.1 (d, J = 21.6 Hz, CH × 2), 106.0 (dd, J = 7.2, 4.1 Hz, CO), 106.3 (dd, J = 7.2, 4.2 Hz, CO), 121.5–121.9 (CH × 2), 143.9 (dd, J = 37.2, 13.4 Hz, CBr), 146.4 (dd, J = 37.9, 13.4 Hz, CBr), 148.0–148.4 (m, CF), 150.5–150.8 (m, CF), 150.9 (dd, J = 7.6, 3.0 Hz, CF), 151.2 (dd, J = 7.5, 2.8 Hz, CF), 167.7 (CO).ESI-TOF MS (m/z): 500.89 (M + 1). HMRS: (ESI-TOF) calcd for C16H10O4F4NaBr2 522.8755 [M + Na]+, found 522.8780.
2-(2-Bromo-4-methylphenoxy)ethyl 2-(2-bromo-4-methylphenoxy)acetate (2d). Following the general procedure, compound (1d) (0.30 g, 1.30 mmol), celite (1.2 g) and PCC (0.56 g, 2.60 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (5:1)) compound (2d) was obtained as a white solid (0.139 g, 47%). 1H NMR (400 MHz, CDCl3) δ: 2.24 (s, 3H, CH3), 2.28 (s, 3H, CH3), 4.20 (t, J = 4.7 Hz, 2H, CH2), 4.58 (t, J = 4.7 Hz, 2H, CH2), 4.73 (s, 2H, CH2COOR), 6.75 (dd, J = 13, 8 Hz, 2H ArH), 6.94 (dd, J = 8, 2 Hz, 1H, ArH), 7.03 (dd, J = 8, 2 Hz, 1H, ArH), 7.35–7.37 (m, 2H, ArH). 13C NMR (101 MHz, CDCl3) δ: 20.3 (CH3), 20.3 (CH3), 63.4 (CH2), 66.6 (CH2), 67.4 (CH2), 112.2 (CBr), 112.5 (CBr), 114.1 (CH), 114.2 (CH), 129.0 (CH), 129.0 (CH), 132.6 (CO), 133.1 (CO), 134.0 (CH), 134.1 (CH), 152.4 (C), 152.8 (C), 168.6 (CO).ESI-TOF MS (m/z): 456.14 (M + 1).
2-((1-Bromonaphthalen-2-yl)oxy)ethyl 2-((1-bromonaphthalen-2-yl)oxy)acetate (2e). Following the general procedure, compound (1e) (0.30 g, 1.12 mmol), celite (0.97 g) and PCC (0.48 g, 2.24 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (5:1)) compound (2e) was obtained as a white solid (0.22 g, 75%). 1H NMR (400 MHz, CDCl3) δ: 4.37 (t, J = 4.6 Hz, 2H), 4.67 (t, J = 4.6 Hz, 2H), 4.90 (s, 2H, CH2COOR), 7.17 (d, J = 9 Hz, 2H), 7.36–7.44 (m, 2H), 7.53–7.60 (m, 2H), 7.66 (dd, J = 14; 9 Hz, 2H), 7.76 (dd, J = 11, 9 Hz, 2H), 8.22 (d, J = 8.6 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 63.7 (CH2), 67.2 (CH2), 68.3 (CH2), 110.4 (C), 110.5 (C), 115.3 (CH), 115.8 (CH), 124.9 (CH), 125.0 (CH), 126.5 (2 × CH), 127.9 (CH), 127.9 (CH), 128.1 (CH), 128.2 (CH), 129.1 (CH), 129.2 (CH), 130.4 (C), 130.5 (C), 133.2 (C), 152.5 (C), 152.9 (C), 168.8 (CO).ESI-TOF MS (m/z): 528.96 (M + 1).
2-((2-Bromopyridin-3-yl)oxy)ethyl 2-((2-bromopyridin-3-yl)oxy)acetate (2f). Following the general procedure, compound (1f) (0.30 g, 1.38 mmol), celite (1.2 g), and PCC (0.59 g, 2.75 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (1:2)) compound (2f) was obtained as a white oil (0.166 g, 56%). 1H NMR (400 MHz, CDCl3) δ: 4.24 (t, J = 4.5 Hz, 2H), 4.62 (t, J = 4.5 Hz, 2H) 4.78 (s, 2H, CH2COOR), 7.10–7.13 (m, 3H, ArH), 7.18–7.22 (m, 1H, ArH), 7.96–8.00 (m, 2H, ArH).13C NMR (101 MHz, CDCl3) δ: 63.1 (CH2), 66.0 (CH2), 67.0 (CH2), 120.3 (CH), 120.7 (CH), 123.4 (CH), 123.6 (CH), 133.2 (CBr), 133.2 (CBr), 142.2 (CH), 142.7 (CH), 151.5 (C), 151.8 (C), 167.7 (CO).
ESI-TOF MS (m/z): 430.92 (M + 1).
2-(2-Bromo-4-cyanophenoxy)ethyl 2-(2-bromo-4-cyanophenoxy)acetate (2g). Following the general procedure, compound (1g) (0.30 g, 1.24 mmol), celite (1.07 g) and PCC (0.54 g, 2.49 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (5:1)) compound (2g) was obtained as a white solid (0.177 g, 59%). 1H NMR (400 MHz, CDCl3) δ: 4.30 (t, J = 4.5 Hz, 2H, CH2), 4.66 (t, J = 4.4 Hz, 2H, CH2), 4.84 (s, 2H, CH2COOR), 6.83 (d, J = 8.5 Hz, 1H, ArH), 6.90 (d, J = 8.5 Hz, 1H, ArH), 7.51 (d, J = 8.5 Hz, 1H, ArH), 7.6 (d, J = 8.6 Hz, 1H, ArH), 7.84 (d, J = 5.7 Hz, 1H, ArH). 13C NMR (101 MHz, CDCl3) δ: 63.0 (CH2), 65.9 (CH2), 67.0 (CH2), 106.2 (CBr), 106.7 (CBr), 112.9 (CH), 113.1 (CH), 117.5 (C), 117.6 (C), 133.0 (CH), 133.3 (CH), 137.1 (CH), 137.3 (CH), 157.8 (C), 158.2 (C), 167.3 (CO).ESI-TOF MS (m/z): 478.92 (M + 1).
2-((3-Bromonaphthalen-2-yl)oxy)ethyl 2-((3-bromonaphthalen-2-yl)oxy)acetate (2h). Following the general procedure, compound (1h) (0.17 g, 0.64 mmol), celite (0.60 g), and PCC (0.30 g, 1.30 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (5:1)) compound (2h) was obtained as a white solid (0.11 g, 66%). 1H NMR (400 MHz, CDCl3) δ: 4.38 (t, J = 4.6 Hz, 2H, CH2), 4.57 (t, J = 4.5 Hz, 2H, CH2), 4.90 (s, 2H, CH2COOR), 7.17 (d, J = 9 Hz, 2H, ArH), 7.36–7.44 (m, 2H, ArH), 7.53–7.6 (m, 2H, ArH), 7.67 (dd, J = 14, 8 Hz, 2H, ArH), 7.76 (dd, J = 10.6, 8.5 Hz, 2H, ArH), 8.22 (d, J = 8.6 Hz, 2H, ArH). 13C NMR (101 MHz, CDCl3) δ: 63.7 (CH2), 67.2 (CH2), 68.3 (CH2), 110.4 (C), 110.5 (C), 115.3 (CH), 115.8 (CH), 125.0 (CH), 125.0 (2 × CH), 126.5, 127.9 (CH), 128.0 (CH), 128.1 (CH), 128.2 (2 × CH), 129.1 (CH), 129.2 (CH), 130.4 (C), 130.5 (C), 133.2 (C), 152.5 (C), 152.9 (C), 168.8 (CO).ESI-TOF MS (m/z): 528.97 (M + 1).
2-(2-Bromo-3-methoxyphenoxy)ethyl 2-(2-bromo-3-methoxyphenoxy)acetate (2i). Following the general procedure, compound (1i) (0.30 g, 1.22 mmol), celite (1.06 g) and PCC (0.53 g, 2.45 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (2i) was obtained as a white solid (0.136 g, 46%). 1H NMR (400 MHz, CDCl3) δ: 3.87 (s, 3H, OMe), 3.89 (s, 3H, OMe), 4.23 (t, J = 4.8 Hz, 2H, CH2), 4.60 (t, J = 4.8 Hz, 2H, CH2), 4.76 (s, 2H, CH2COOR), 6.47 (d, J = 8 Hz, 1H, ArH), 6.52–6.60 (m, 3H, ArH), 7.12 (t, J = 8 Hz, 1H, ArH), 7.20 (t, J = 8 Hz, 1H, ArH). 13C NMR (101 MHz, CDCl3) δ: 56.6 (2 × OMe), 63.3 (CH2), 66.4 (CH2), 67.2 (CH2), 101.9 (CBr), 102.0 (CBr), 105.4 (CH), 105.8 (CH), 106.2 (CH), 106.4 (CH), 128.3 (CH), 128.4 (CH), 155.7 (C), 156.2 (C), 157.4 (C), 157.4 (C), 168.5 (CO).ESI-TOF MS (m/z): 488.95 (M + 1).
2-(Phenoxy)ethyl 2-(phenoxy)acetate (2j). Following the general procedure, compound (1j) (0.24 g, 1.76 mmol), celite (1.52 g) and PCC (0.76 g, 3.52 mmol) were added to DCM (5 mL) in a round-bottom-flask and allowed to react as described above. After purification by silica gel chromatography (Hex
:EtOAc (2:1)) compound (2j) was obtained as a white solid (0.161 g, 67%). 1H NMR (400 MHz, CDCl3) δ: 4.2 (t, J = 4.7 Hz, 2H, CH2), 4.58 (t, J = 4.7 Hz, 2H, CH2), 4.68 (s, 2H, CH2), 6.92 (d, J = 8 Hz, 4H, ArH), 7.00 (t, J = 7 Hz, 2H, ArH), 7.29 (dd, J = 19, 8 Hz, 4H, ArH). 13C NMR (101 MHz, CDCl3) δ: 63.6 (CH2), 65.4 (CH2), 65.7 (CH2), 114.7 (2 × CH), 114.8 (2 × CH), 121.4 (CH), 121.9 (CH), 129.7 (2 × CH), 129.7 (2 × CH), 157.9 (C), 158.4 (C), 169.0 (CO). ESI MS (m/z): 272.90 (M).
:1) was added to the crude product. This mixture was filtered over a silica pad under vacuum. The solvent was evaporated under reduced pressure. After purification by silica gel chromatography compounds (2a), was obtained as a white solid (0.109 g, 61%).Acknowledgements
The authors gratefully acknowledge the following funding sources: the INMOLFARM – Molecular Innovation and Drug Discovery (ALENT-07-0224-FEDER-001743) of the FEDER-INALENTEJO program including, a PhD grant to HV and research grants to PB and AG. EPC thanks the Fundação para a Ciência e a Tecnologia (FCT) for a post-doctoral research fellowship (SFRH/BPD/72182/2010). We acknowledge project LADECA (ALENT-07-0262-FEDER-001878) for the acquisition of the high-field NMR 400 MHz Bruker Avance III machine. The Foundation for Science and Technology (FCT) in Portugal is acknowledged for funding through the strategic project PEst-OE/QUI/UI0619/2014. We thank Dr Ricardo Mendonça (Hovione, Loures, Portugal) for useful insights and discussions on the possible oxidative esterification reaction. We also acknowledge Dr Kerry Gilmore from MPI for Colloids and Interfaces in Potsdam, Germany, for facilitating HRMS analysis of our compounds at the University of Potsdam.Notes and references
- J. Otera and J. Nishikido, Esterification: methods, reactions, and applications, Wiley – VCH, Weinheim, 2nd edn, 2010 Search PubMed.
- S. Tang, J. Yuan, C. Liu and A. Lei, Dalton Trans., 2014, 43, 13460–13470 RSC.
- (a) J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS; (b) R. C. Larock, Comprehensive organic transformations: a guide to functional group preparations, Wiley – VCH, New York, 2nd edn, 1999 Search PubMed; (c) E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78 CrossRef CAS PubMed.
- (a) S. D. Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef PubMed; (b) J. H. Espenson, Z. Zhu and T. H. Zauche, J. Org. Chem., 1999, 64, 1191–1196 CrossRef CAS; (c) R. Gopinath and B. K. Patel, Org. Lett., 2000, 2, 577–579 CrossRef CAS PubMed; (d) B. E. Maki and K. A. Scheidt, Org. Lett., 2008, 10, 4331–4334 CrossRef CAS PubMed; (e) S. Gaspa, A. Porcheddu and L. De Luca, Org. Lett., 2015, 17, 3666–3669 CrossRef CAS PubMed.
- (a) W. J. Tishchenko, Russ. Phys. Chem., 1906, 38, 355 Search PubMedL. Claisen, Ber. Dtsch. Chem. Ges., 1887, 20, 646–650 CrossRef; (b) O. P. Törmäkangas and A. M. P. Koskinen, Recent Res. Dev. Org. Chem., 2001, 5, 225–255 Search PubMed; (c) T. Seki, T. Nakajo and M. Onaka, Chem. Lett., 2006, 35, 824–829 CrossRef CAS; (d) J. March, Advanced Organic Chemistry; Reactions, Mechanisms and Structure, John Wiley & Sons Inc, New Jersey, New York, 4th edn, 1992 Search PubMed; (e) B. P. Mundy, M. G. Ellend and F. G. Favaloro Jr, Name Reactions and Reagents in Organic Synthesis, John-Wiley & Sons, Inc, New Jersey, 2nd edn, 2005. Search PubMed And for some interesting recent applications see: (f) S. P. Curran and S. J. Connon, Angew. Chem., Int. Ed., 2012, 51, 10866–10870 CrossRef CAS PubMed; (g) M. K. Barman, A. Baishya and S. Nembenna, J. Organomet. Chem., 2015, 785, 52–60 CrossRef CAS; (h) T. Werner and J. Koch, Eur. J. Org. Chem., 2010, 6904–6907 CrossRef CAS.
- (a) T. Suzuki, T. Matsuo, K. Watanabe and T. Katoh, Synlett, 2005, 1453–1455 CrossRef CAS; (b) C. Liu, S. Tang, L. Zheng, D. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 5662–5666 CrossRef CAS PubMed; (c) S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5139–5143 CrossRef CAS PubMed; (d) C. Liu, J. Wang, L. Meng, Y. Deng, Y. Li and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 5144–5148 CrossRef CAS PubMed; (e) R. Ray, R. D. Jana, M. Bhadra, D. Maiti and G. K. Lahiri, Chem.–Eur. J., 2014, 20, 15618–15624 CrossRef CAS PubMed; (f) J. Xia, A. Shao, S. Tang, X. Gao, X. Gao and A. Lei, Org. Biomol. Chem., 2015, 13, 6154–6157 RSC.
- (a) E. J. Corey and J. W. Suggs, Tetrahedron Lett., 1975, 31, 2647–2650 CrossRef . For general information on the synthetic applications of this reagent see: ; (b) W. Carruthers and I. Coldham, Modern methods of organic synthesis, Cambridge University Press, Cambridge, U.K, 4th edn, 2004 Search PubMed; (c) W. Carruthers, Some modern methods of organic synthesis, Cambridge University Press, Cambridge, U.K, 3rd edn, 1987 Search PubMed.
- J. C. Craig and E. C. Horning, J. Org. Chem., 1960, 25, 2098–2102 CrossRef.
- M. B. Yunker and B. J. C. S. Fraser-Reid, Chem. Commun., 1975, 61–62 RSC.
- L. Ermolenko, N. A. Sasaki and P. Potier, Synlett, 2001, 10, 1565–1566 CrossRef.
- Z. Guo, R. Padmakumar, R. I. Hollingsworth, K. V. Radhakrishnan, M. V. Nandakumar and B. Fraser-Reid, Synlett, 2003, 7, 1067–1069 Search PubMed.
- M. Hunsen, Tetrahedron Lett., 2005, 46, 1651–1653 CrossRef CAS.
- (a) V. Piccialli, S. Zaccaria, G. Oliviero, S. D'Errico, V. D'Atri and N. Borbone, Eur. J. Org. Chem., 2012, 4293–4305 CrossRef CAS; (b) S. Bali, M. A. Tofanelli, R. D. Ernst and E. M. Eyring, Biomass Bioenergy, 2012, 42, 224–227 CrossRef CAS; (c) E. S. Gould, Coord. Chem. Rev., 1994, 135, 651–684 CrossRef; (d) R. C. Mehrotra, P. C. Bharara and K. N. Mahendra, Transition Met. Chem., 1977, 2, 161–163 CrossRef CAS.
- (a) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed; (b) A. K. Oyelere, Curr. Top. Med. Chem., 2010, 10, 1359–1360 CrossRef CAS PubMed; (c) E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed.
- H. Viana, PhD Dissertation, University of Evora, 2016.
- P. Bacalhau, A. San Juan, C. S. Marques, D. Peixoto, A. J. Burke, A. T. Caldeira and M. R. Martins, Neurodegener. Dis., 2015, 15(suppl. 1), 741 Search PubMed; P. Bacalhau, A. San Juan, C. S. Marques, D. Peixoto, A. Goth, C. Guarda, M. Silva, S. Arantes, A. T. Caldeira, M. R. Martins and A. J. Burke, Bioorg. Chem., 2016, 67, 1–8 CrossRef CAS PubMed.
- G. Ellman, K. Courtney, V. Andres and R. Featherstone, Biochem. Pharmacol., 1961, 7, 88 CrossRef CAS PubMed.
- N. Yamamoto, M. Komatsu, Y. Suzuki, K. Kawano, T. Kimura, K. Ito, S. Nagato, Y. Norimine, T. Niidome, T. Teramoto, Y. Iimura and S. Hatakeyama, EP1099692A1, 2001.
- N. Lachance, C. S. Li, J.-P. Leclerc and Y. K. Ramtohul, WO2008/128335A1, 2008.
- PubChem data base, https://pubchem.ncbi.nlm.nih.gov/compound/58241908#section=Top.
- PubChem data base, https://pubchem.ncbi.nlm.nih.gov/compound/28602536#section=Top.
- L. F. Tietze and F. Lotz, Eur. J. Org. Chem., 2006, 20, 4676–4684 CrossRef.
- E. Marsault, K. Benakli, H. Hoveyda, M. Peterson, S. Beaubien, L. Oullet, C. St-Louis and S. Beauchemin, Patent US9181298 B2, 2015.
- L. F. Tietze, T. Hungerland, A. Düfert, I. Objartel and D. Stalke, Chem.–Eur. J., 2012, 18, 3286–3291 CrossRef CAS PubMed.
- E. Brenna, F. Cannavale, M. Crotti, F. Parmeggiani, A. Romagnolo, F. Spina and G. C. Varese, J. Mol. Catal. B: Enzym., 2015, 116, 83–88 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra07547a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |