DOI:
10.1039/C6RA07452A
(Paper)
RSC Adv., 2016,
6, 54013-54019
Green synthesis of polyureas from CO2 and diamines with a functional ionic liquid as the catalyst†
Received
22nd March 2016
, Accepted 27th May 2016
First published on 31st May 2016
Abstract
A series of ionic liquids (P4,4,4,6BF4, P4,4,4,6Triz, as well as the newly prepared anion dual-functionalized amino-triz IL P4,4,4,6ATriz, etc.) were prepared, and their catalytic performance was tested in the synthesis of polyureas from CO2 and diamines. Under the optimized reaction conditions, good to excellent yields of various polyureas were achieved with different diamines over P4,4,4,6ATriz catalyst. It can be found that the catalytic performance is essentially consistent with the basicity of ILs (as determined by TPD method). The solid products were characterized extensively by 13C NMR, FT-IR, XRD, DSC and TGA. From these results, it could be concluded that the solid products based on diamines and CO2 have the polyurea structure with the urea linkage and connected by hydrogen bonds, which resulted in their high resistance to solvents and excellent thermal stability.
1. Introduction
As a renewable, abundant, cheap, and non-toxic carbon resource, transformation of CO2 to other useful carbon compounds has attracted significant interest.1,2 Among them, urea derivatives are widely used as intermediates in the synthesis of pharmaceuticals and agricultural chemicals.3–5 Meanwhile, they can also be regarded as potential precursors of carbamates and isocyanates, which are useful raw materials of polyurethanes.6 Although the 1,3-disubstituted ureas and cyclic ureas are obtained in moderate to excellent yields from monoamine7–12 and ethylene diamine,13 respectively, there have been limited reports of polyurea derivative synthesis directly from CO2 and diamines. Polyurea is a new kind of polymer containing the urea functional group (–NH–CO–NH–), which shows good resistance to solvents, far superior thermal properties and much higher melting points than the commonly used polymers. Its fast reactivity and relative moisture insensitivity make it useful for coatings on large surface area projects, such as secondary containments, manhole and tunnel coatings, tank liners, and truck bed liners. They can also be used for spray molding and armor. Polyurea is generally produced from toxic isocyanates and amino compounds in the industrial processes.14–16 Nowadays, replacement of these hazardous reagents in chemical processes is one of the main goals in green chemistry. Clearly, the direct use of CO2 as a building block, e.g. carbonyl source, in the production of polyureas should be an ideal choice. However, this route usually needs stoichiometric toxic catalysts (such as diphenyl phosphate, phosphorus chlorides or N-acylphosphoramidites).17,18 Recently, polyurea was successfully synthesized via the reaction of CO2 with diamines under catalyst-free conditions,19–22 however, the relatively high pressure (i.e. 12 MPa) and long time limit its practical application.
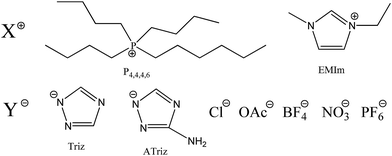
Room-temperature ionic liquids (ILs) as alternative reaction media and catalytic materials have been attracted a lot of interest due to their intrinsic physicochemical properties, such as the negligible vapor pressure, non-flammability and unlimited combinations of cations and anions. Moreover, various functionalized ILs, incorporating different functional groups into the ILs structure, have been explored and used as solvents or catalysts in chemical processes.23–25 Herein, a series of amine-functionalized ILs (Scheme 1) were prepared and their catalytic performance for polyureas synthesis from diamines and CO2 at relatively lower pressure was tested. The influences of the structure and property of ILs, the scope of application and the reusability of ILs for polyureas synthesis were also investigated.
 |
| | Scheme 1 Structures and abbreviations of the cations and anions used in this study. | |
2. Experimental
2.1 General procedure for the preparation of amine-functionalized ionic liquids
ILs used in this study including 1-ethyl-3-methylimidazolium chloride (EMImCl), 1-ethyl-3-methylimidazolium tetrafluoroborate (EMImBF4), 1-ethyl-3-methylimidazolium hexafluorophosphate (EMImPF6), 1-ethyl-3-methylimidazolium nitrate (EMImNO3), 1-ethyl-3-methylimidazolium acetate (EMImOAc), were synthesized by our lab with 98–99% of purity. Hexyltributylphosphonium triazole (P4,4,4,6Triz) and hexyltributylphosphonium aminotriazole (P4,4,4,6ATriz) were prepared by the neutralization of hexyltributylphosphonium hydroxide (P4,4,4,6OH) and 1,2,4-triazole or 3-amino-1,2,4-triazole according to literature methods.26,27 The structures of these ILs were confirmed by NMR spectroscopy. 1H NMR and 13C NMR spectra were recorded on a Bruker AMX FT 400 MHz NMR spectrometer (Fig. S1†). The ILs were dried several hours at 80 °C to 100 °C under a vacuum to separate the ILs from volatile byproducts and humidity. Thus, the amount of water in all used ILs is less than 400 ppm.
2.2 General procedure for the reaction of CO2 and amines
All reactions were carried out in a 90 mL stainless steel autoclave with a magnetic stirrer. As an example, 10 mmol of 1,6-hexamethylenediamine (HDA), 3 mL N-methyl-2-pyrrolidone (NMP) and 0.2 g P4,4,4,6ATriz were charged in the reactor, and the reactor was saturated with CO2 under a pressure of 4 MPa at room temperature. The reaction proceeded at 170 °C for 8 h. After the reaction, the autoclave was cooled down to room temperature and carefully depressurized. The products were washed with water and weighed to determine the isolated yields.
2.3 Characterization of the catalysts and products
The CP/MAS 13C NMR spectra was recorded on a Bruker AVANCE II WB 400 spectrometer equipped with a 4 mm standard bore CP/MAS probe head. Fourier transform infrared spectroscopy (FT-IR) transmission data were collected from pressed catalyst disk made with KBr in the range of 4000–400 cm−1 with a Nicolet 5700 FT-IR. X-ray diffraction (XRD) measurements were carried out on a Siemens D/max-RB powder X-ray diffractometer. Differential scanning calorimetry (DSC) experiments were carried out on a Mettler-Toledo DSC822e calorimeter with heating and cooling-rates of 10 °C min−1 from 0 °C up to 300 °C under N2 flow. Thermal analysis (TGA) was carried out using a METTLER TG1 model. Samples were heated from room temperature to 800 °C at a heating rate of 10 °C min−1 under a nitrogen flow of 50 mL min−1. Temperature-programmed desorption (TPD) was performed using a TPD flow system equipped with a thermal conductivity detector (TCD). The quantitative analysis for CO2 desorption is calculated based on the integration of the corresponding TPD traces, preliminarily calibrated by the injection of pure CO2 pulses.
3. Results and discussion
3.1 Polyureas syntheses from diamines and CO2
In the preliminary study, 1,6-hexanediamine (HDA) was selected as a model substrate to test this protocol as shown in Table 1. It is worthy to mention that carbamate salt was the only by-product found after the reaction, which could be removed by adding water. The insoluble solid products obtained from HDA with CO2 in the absence of catalyst (entry 1) were analyzed with CP/MAS 13C NMR (Fig. S2†), and the characterization peak at 159.7 ppm indicating the formation of carbonyl in the urea linkage19 When HDA is placed under CO2 pressure, carbamate salt is formed spontaneously, while the further transformation of carbamate salts into ureas normally requires high temperature and pressure, or the presence of catalysts (such as NEt3, or DBU).28,29 Herein, six different anions combined with the [EMIm] cation were employed for the synthesis of polyureas, and the yield of polyurea–HDA could be greatly improved when ILs used as catalysts (entries 2–7). According to the results, it could be found that the anion has a strong impact on the catalytic activities; the effect of the anion on the yield of polyurea–HDA follows the order: [OAc] > [Triz] > [BF4] > [NO3] > [PF6] > [Cl]. The EMImOAc gives high activity than EMImPF6 and EMImCl could be due to the basicity of [OAc] is much stronger than PF6 and Cl. However, EMImOAc is easily decomposed at about 170 °C, which would limit the recycle of IL. Since we note that the catalytic activity of ILs might relate to the basicity of anion, we described a new strategy for significantly increasing the basicity of ILs by the introduction of an ammonium group on the triz-based anion. As expected, the yield of polyurea–HDA increased to 88% (entry 8). The effect of phosphonium based amine-functionalized ILs with different anions (BF4−, Triz, ATriz) on the reaction was also investigated (entries 7 and 8). It was found that P4,4,4,6ATriz showed the best performance in the synthesis of polyurea–HDA, and the yield of polyurea–HDA could reach 96%. From these results, it could be found that both the cation and anion of the investigated ILs have a strong impact on the catalytic activities.
Table 1 Syntheses of polyurea–HDA from HDA and CO2 with different catalystsa

|
| Entry |
Catalyst |
Yieldb (%) |
| Reaction conditions: 10 mmol HDA, 3 mL NMP, 170 °C, 8 h, 4 MPa, 90 mL autoclave. Isolated yield based on charged HDA. |
| 1 |
None |
27 |
| 2 |
EMImCl |
40 |
| 3 |
EMImPF6 |
59 |
| 4 |
EMImNO3 |
61 |
| 5 |
EMImBF4 |
79 |
| 6 |
EMImTriz |
82 |
| 7 |
EMImOAc |
83 |
| 8 |
EMImATriz |
88 |
| 9 |
P4,4,4,6BF4 |
68 |
| 10 |
P4,4,4,6Triz |
89 |
| 11 |
P4,4,4,6ATriz |
96 |
| 12 |
P4,4,4,6ATriz-reused1st |
95 |
| 13 |
P4,4,4,6ATriz-reused2nd |
92 |
| 14 |
P4,4,4,6ATriz-reused3rd |
93 |
The reusability of ILs is a key parameter in determining their suitability in practical applications. Firstly, the thermal stability of P4,4,4,6ATriz (Fig. S3†) was tested by thermal gravimetric analyzer (TGA). The initial decomposition temperature was more than 250 °C, indicating that P4,4,4,6ATriz has high thermal stability and no decomposition of P4,4,4,6ATriz was observed under our experimental conditions. Therefore, the P4,4,4,6ATriz in the filtrate was recovered by removing water under vacuum, and reused without any processing. >92% yields were obtained after 3 times recycle (entries 12–14), suggesting the catalyst was stable and the catalytic activity could be essentially preserved. Besides, there was almost no difference in the IR spectrum between fresh and recovered P4,4,4,6ATriz, which also provide an evidence for the stability of the employed catalyst (Fig. S4†).
3.2 Effect of reaction conditions
Subsequently, the effects of the reaction temperature, time, CO2 pressure and amount of P4,4,4,6ATriZ on the yield of polyurea–HDA were investigated (Table 2). The effect of temperature on the reaction was investigated in the range of 150–190 °C. The yield of polyurea–HDA increased rapidly with increasing the reaction temperature and the yield reached a maximum at 170 °C, i.e. 96% (entries 1–3). However, with the further increase of reaction temperature the yield of polyurea–HDA declined rapidly. This may be due to that it was an exothermic reaction, and high temperature will lead to the reaction shifted to left.30 By increasing the reaction times, the yield of polyurea–HDA increased smoothly and reached a maximum yield after 8 h (entries 2, 4 and 5). It was also found that the polyurea–HDA yield was sensitive to the CO2 pressure (entries 6–8). The yield of polyurea–HDA increased with increasing the pressure up to 4 MPa. The main reason is that at lower pressure, an increase in pressure enhances the reaction rate because CO2 is a reactant and the solubility of CO2 in the reaction phase increased with increasing CO2 pressure. When the pressure is higher than 4 MPa, the concentration of CO2 in the reaction phase is high enough, and therefore, the effect of pressure on the yield is very limited at the higher pressure. Moreover, the amount of ILs played an important role for such reaction (entries 9–11). The yield increased from 63% to 97% when the amount of P4,4,4,6ATriz increased from 0.1 g to 0.2 g. With further increase the catalyst concentration, the yield of the polyurea–HDA showed slightly decreased, which might be due to the much strong interaction between P4,4,4,6ATriz and CO2.
Table 2 Syntheses of polyurea–HDA from HDA and CO2 with different reaction conditionsa

|
| Entry |
Catalyst (mg) |
Temp. (°C) |
Time (h) |
PCO2 (MPa) |
Yieldb (%) |
| Reaction conditions: 10 mmol HDA, 3 mL NMP, 90 mL autoclave. Isolated yield based on charged HDA. |
| 1 |
200 |
150 |
8 |
4 |
67 |
| 2 |
200 |
170 |
8 |
4 |
97 |
| 3 |
200 |
190 |
8 |
4 |
91 |
| 4 |
200 |
170 |
4 |
4 |
54 |
| 5 |
200 |
170 |
12 |
4 |
97 |
| 6 |
200 |
170 |
8 |
2 |
25 |
| 7 |
200 |
170 |
8 |
3 |
74 |
| 8 |
200 |
170 |
8 |
5 |
97 |
| 9 |
100 |
170 |
8 |
4 |
63 |
| 10 |
300 |
170 |
8 |
4 |
93 |
| 11 |
400 |
170 |
8 |
4 |
88 |
3.3 Polyureas syntheses from various diamines and CO2
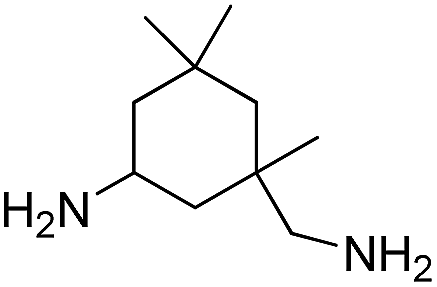
In order to investigate the limitation and scope of this protocol, the reaction between several different aliphatic diamines, such as ethylenediamine (EDA), 1,4-butanediamine (BDA), isophorone diamine (IPDA), 4,4′-diaminodicyclohexyl methane (HMDA), and with CO2 for corresponding polyureas were further tested in the optimized conditions, and the results are given in Table 3. First, the impact of the chain length of linear diamines on the CO2 reaction for corresponding polyureas was tested (entries 1–3). The yields of corresponding polyureas of linear diamines increased rapidly with increasing the chain length. This could be ascribed to the cyclization of short chain amines, and the main product was cycle urea when EDA as reactant. With IPDA and HMDA as the reactants, the yields of the corresponding polyureas were 91% and 90%, respectively (entries 4 and 5). Compared with the linear amines, the cyclic amines were considerably less reactive, which could be ascribed to the high steric hindrance of diamines caused by the cyclic structure. All those results suggested that the polyureas derivatives were successfully formed from aliphatic diamines and CO2.
Table 3 Syntheses of polyurea and urea derivatives with different diaminesa
Then, the reaction of various aromatic diamines and CO2 was also studied (entries 6–9). Under the optimized reaction conditions, no polyureas products could be detected when toluene diamine (TDA) and 4,4-diaminodiphenyl methane (MDA) were used as the reactant. As 1,4-diaminobenzene was used, the yield of the corresponding polyurea was only 22% (entry 8). The inactivity of aromatic diamines for these reactions may result from the π-bond in their molecular structure, which strongly decreases the electron cloud density of the N atom and reduces the ability of N atom combined with proton, i.e. basicity decreased. Aside from their basicity, the weaker nucleophilicity of the aromatic amine was also considered to contribute the low reactive activity. However, 1,3-benzenedimethylamine (MXDA) could be successfully converted to corresponding polyurea and 87% yield was obtained (entry 9). This may be due to MXDA has no aromatic ring attached directly to the N atom.
3.4 Possible reaction mechanism
In order to explore the relationship between the catalytic activity and the IL structure and properties, the interaction between ILs and CO2 was investigated by the TPD technology (Fig. 1). There was no peak was appeared in the desorption curve of P4,4,4,6BF4, which indicated that the interaction between CO2 and P4,4,4,6BF4 is very weak. For P4,4,4,6Triz, two desorption peaks were observed at 59 °C and 120 °C, indicated to two absorption sites corresponding to the P4,4,4,6Triz anion (Fig. S5†).31 To further study the interaction between CO2 and P4,4,4,6Triz, FT-IR analysis were performed to support the results of TPD. A new peak was observed at 1718 cm−1 in the FTIR spectrum after CO2 absorption, attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of carbamate (Fig. S6†). In comparison with that of P4,4,4,6Triz, the desorption capacity of EMImTriz decreased to 0.15 molCO2 per molILs (Table 4), while the temperature of the desorption peaks increased to 79 °C and 151 °C. These results suggest that EMImTriz has stronger basic strength but lesser total basic sites as compared to P4,4,4,6Triz. On the other hand, the P4,4,4,6ATriz showed two broad and strong desorption peaks at about 80–140 °C with a considerable amount of basic sites (0.79 molCO2 per molILs). The relatively high desorption capacity was attributed to the formation of ammonium carbamate salt by CO2 and ammonium ILs. From the preliminary results, it can be conjectured that the strong basicity might be the major reason for the highly catalytic activity of P4,4,4,6Triz. Moreover, the combination effect between cations and anions was also considered to contribute to the high catalytic activity.
O stretch of carbamate (Fig. S6†). In comparison with that of P4,4,4,6Triz, the desorption capacity of EMImTriz decreased to 0.15 molCO2 per molILs (Table 4), while the temperature of the desorption peaks increased to 79 °C and 151 °C. These results suggest that EMImTriz has stronger basic strength but lesser total basic sites as compared to P4,4,4,6Triz. On the other hand, the P4,4,4,6ATriz showed two broad and strong desorption peaks at about 80–140 °C with a considerable amount of basic sites (0.79 molCO2 per molILs). The relatively high desorption capacity was attributed to the formation of ammonium carbamate salt by CO2 and ammonium ILs. From the preliminary results, it can be conjectured that the strong basicity might be the major reason for the highly catalytic activity of P4,4,4,6Triz. Moreover, the combination effect between cations and anions was also considered to contribute to the high catalytic activity.
 |
| | Fig. 1 TPD-CO2 profiles of different ILs supported on glass powder (11 wt%). | |
Table 4 CO2 desorption of different ILs/glass powdera31
| IL |
Desorption capacity (molCO2 per molILs) |
Maximum desorption temperature (°C) |
| Based on TPD. |
| P4,4,4,6BF4 |
0 |
0 |
| EMImTriz |
0.15 |
79, 151 |
| P4,4,4,6Triz |
0.4 |
59, 120 |
| P4,4,4,6ATriz |
0.79 |
80, 136 |
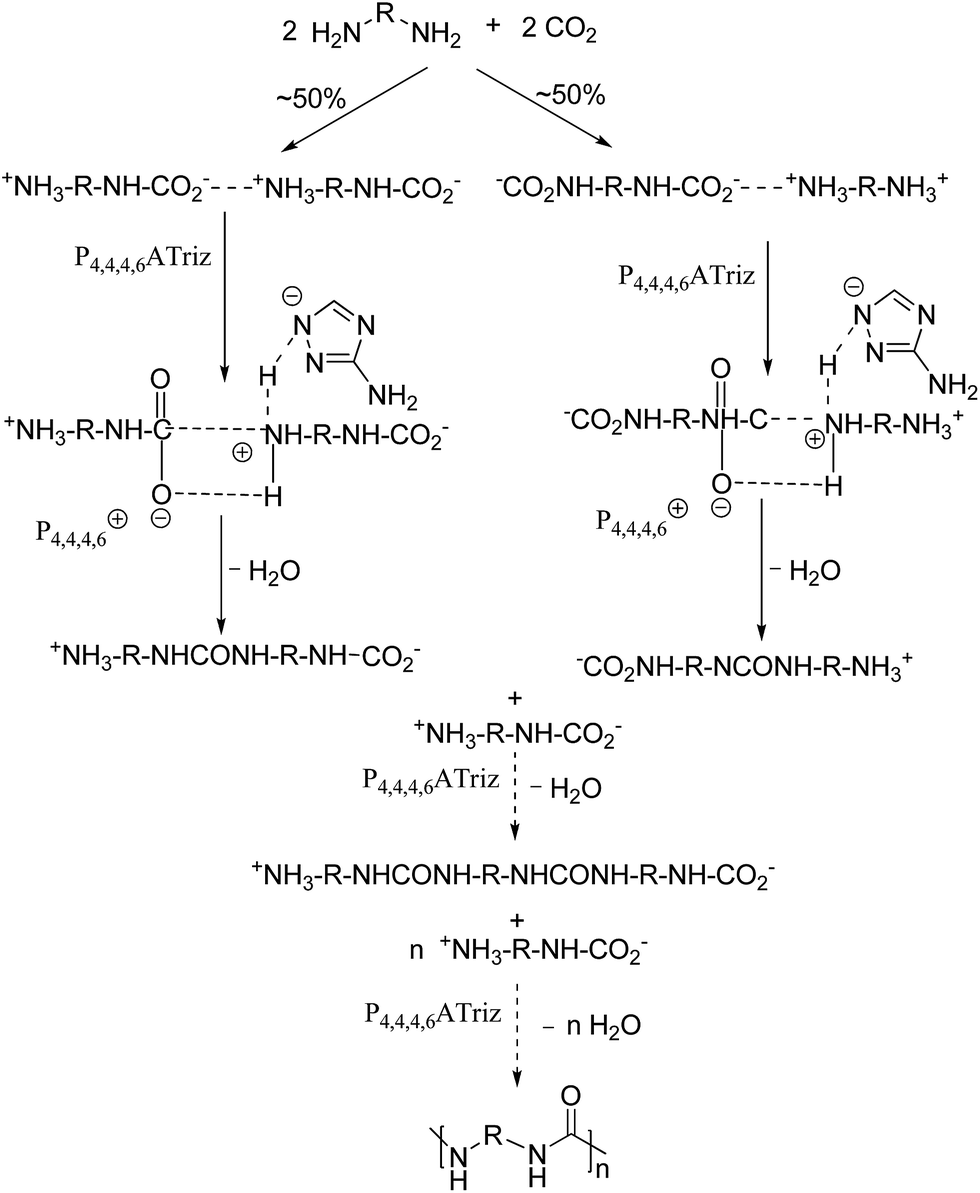
According to the results of the characterization and catalytic performance of P4,4,4,6Triz catalyst, it is known that P4,4,4,6Triz is a basic IL and have strong interaction with CO2. IR result (Fig. S4†) indicated that P4,4,4,6Triz remained its structure after being used. Therefore, we can deduce that P4,4,4,6Triz may complete a catalytic cycle while retaining its structure. Thus, a possible mechanism for the polyureas syntheses from CO2 and diamines over P4,4,4,6Triz was proposed and illustrated in Scheme 2. First, the corresponding carbamate salt intermediate was obtained spontaneously by the reaction of diamines and CO2, the P4,4,4,6Triz catalyst mainly catalyzed the dehydration process of carbamate salt. As a result, the carbamate anion is activated by the hexyltributylphosphonium ion and the carbamate cation is activated by the aminotriazole ion, then urea is formed by the elimination of a water molecule.32 At the same time, P4,4,4,6Triz is regenerated to complete the catalytic reaction cycle. Hence, the polyurea could be gradually formed as mentioned above.
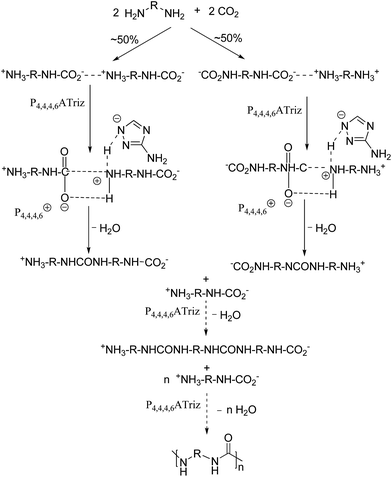 |
| | Scheme 2 Possible reaction mechanism for the polyureas syntheses from CO2 and diamines over P4,4,4,6Triz. | |
3.5 Results of the polyureas characterization
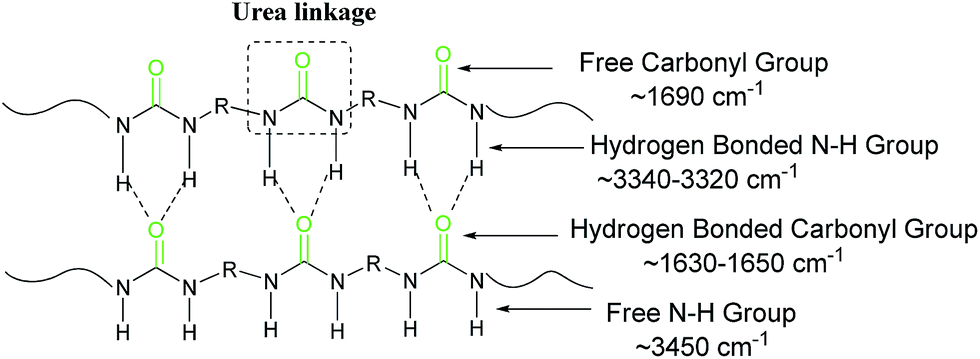
To further study the structure of the solid products, FT-IR spectroscopic analyses (Fig. 2) were performed to support the results of CP/MAS 13C NMR. The peaks at 1577 cm−1 and 1618 cm−1 in polyurea–HDA attribute to the characteristic absorption peaks of carbonyl CO and CO⋯N–H, respectively, suggesting urea derivatives were generated in the products.33,34 Compared to free CO peak (∼1690 cm−1), there is a 72 cm−1 shift in the peak position which indicates fairly strong hydrogen bonding in this compound (Fig. 3). Moreover, the peak for N–H stretch of polyurea–HDA compared with free N–H vibration (∼3450 cm−1) shifted to lower wavenumber 3331 cm−1, which further confirmed the existence of hydrogen bonds in polyurea–HDA. Similar FT-IR spectras were also observed for the solid products obtained from HMDA, IPDA and MXDA with CO2. From these two results, it could be conjectured that the solid product based on diamines and CO2 has the polyureas structure with the urea linkage and connected by the hydrogen bonds. The network structures of these polymers caused their high resistance to solvents. For example, because the polyurea derivatives synthesized in this work were completely insoluble in conventional solvents, the measurement of the molecular weight and distributions become difficult via the traditional methods, such as GPC or MS.
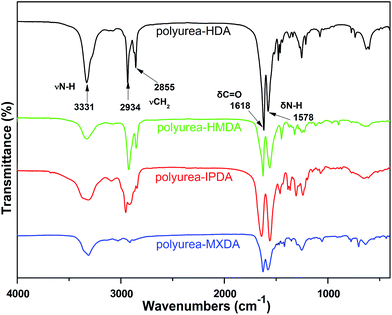 |
| | Fig. 2 FT-IR spectrum of the solid products of diamines reaction with CO2. | |
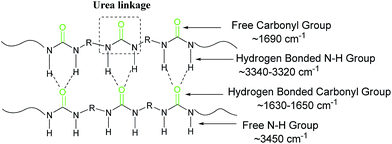 |
| | Fig. 3 The network structure of the polyureas and band assignments for the polyurea CO, N–H stretching modes. | |
The thermal properties of these polyurea derivatives were characterized with DSC and TGA, respectively. The DSC curves (Fig. 4) showed that all the polyureas displayed glass transition behavior during the heating process, but only the polyurea–HDA exhibited a melting point (278 °C). Compared with short chain polyurea–BDA, the long chain polyurea–HDA has a melt state because the long chain carbon structures increased the flexibility of the polymers and then decreased the melting temperature.21
 |
| | Fig. 4 DSC curves of the polyureas. | |
Furthermore, the thermal stability was checked by TGA, where the degradation of the sample can be monitored (Fig. 5). The initial decomposition temperatures are more than 200 °C and the maximum decomposition temperatures are more than 320 °C, indicating that these polyureas have high thermal stability. For the polyurea–BDA, two stage of weight loss could be discovered in TGA curve during the heating process, and the complete decomposition of polyurea–BDA was at about 500 °C. Polyurea–HDA displayed a similar TGA curve with polyurea–BDA, except the initial decomposition temperature shifted to higher temperature (300 °C), demonstrating that the thermal stability increased with the chain length. From the preliminary results, it could be also found that aliphatic polyurea systems have a high decomposition temperature, higher than the aromatic polyurea systems. However, for the polyurea–MXDA, there was approximately 30% weight residual after 800 °C, meaning that polyurea–MXDA may be regarded as potential precursors of carbon materials.
 |
| | Fig. 5 TGA curves of the polyureas. | |
XRD analyses (Fig. 6) indicated that all the polyureas have the semi-crystalline structure except for the polyurea–IPDA. The less flexibility and large hindrance of the cyclic structure with side chain substituent may be the major reason for the lower crystallinity in comparison with other polyureas.
 |
| | Fig. 6 XRD patterns of the polyureas. | |
4. Conclusions
The polyureas were successfully synthesized from diamines and CO2 with a functional ionic liquid (P4,4,4,6ATriz) under mild conditions. TPD result indicated that there was a strong interaction between P4,4,4,6ATriz and CO2, further investigation on the mechanistic pathway will be necessary to clarify the details of this fascinating and important catalytic reaction. The synthesized polyureas exhibited exceptional properties, such as high resistance to solvents and excellent thermal stability, which were caused by their special net structures.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21173240).
Notes and references
- W. Leitner, Angew. Chem., Int. Ed., 1995, 34, 2207–2221 CrossRef CAS.
- A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- K. Matsuda, Med. Res. Rev., 1994, 14, 271–301 CrossRef CAS PubMed.
- F. Bigi, R. Maggi and G. Sartori, Green Chem., 2000, 2, 140–148 RSC.
- G. Bartolo, G. Salerno, R. Mancuso and M. Costa, J. Org. Chem., 2004, 69, 4741–4750 CrossRef PubMed.
- D. Kim, E. Huh, S. Park, L. Nguyen, M. Nguyen, H. Kim, M. Cheong and D. Nguyen, Adv. Synth. Catal., 2010, 352, 440–446 CrossRef CAS.
- F. Shi, Y. Deng, T. SiMa, J. Peng, Y. Gu and B. Qiao, Angew. Chem., Int. Ed., 2003, 42, 3257–3260 CrossRef CAS PubMed.
- D. Niu, L. Zhang, L. Xiao, Y. Luo and J. Lu, Appl. Organomet. Chem., 2007, 21, 941–944 CrossRef CAS.
- J. Fournier, C. Bruneau, P. H. Dixneuf and S. Lécolier, J. Org. Chem., 1991, 56, 4456–4458 CrossRef CAS.
- T. Jiang, X. Ma, Y. Zhou, S. Liang, J. Zhang and B. Han, Green Chem., 2008, 10, 465–469 RSC.
- Y. Shim, J. Lee, J. Im, D. Mukherjee, D. Nguyen, M. Cheong and H. Kim, Phys. Chem. Chem. Phys., 2011, 13, 6197–6204 RSC.
- D. Nguyen, J. Cho, S. Shin, D. Mishra and Y. Kim, ACS Sustainable Chem. Eng., 2016, 4, 451–460 CrossRef CAS.
- B. M. Bhanage, S. Fujita, Y. Ikushima and M. Arai, Green Chem., 2003, 5, 340–342 RSC.
- J. Mattia and P. Painter, Macromolecules, 2007, 40, 1546–1554 CrossRef CAS.
- T. Choi, J. Weksler, A. Padsalgikar and J. Runt, Polymer, 2009, 50, 2320–2327 CrossRef CAS.
- A. Sanchez-Ferrer, D. Rogez and P. Martinoty, Macromol. Chem. Phys., 2010, 211, 1712–1721 CrossRef CAS.
- N. Yamazaki, T. Tomioka and F. Higashi, J. Polym. Sci., Part C: Polym. Lett., 1976, 14, 55–57 CrossRef CAS.
- G. Rokicki, Macromol. Chem. Phys., 1988, 189, 2513–2520 CrossRef CAS.
- C. Wu, J. Wang, P. Chang, H. Cheng, Y. Yu, Z. Wu, D. Dong and F. Zhao, Phys. Chem. Chem. Phys., 2012, 14, 464–468 RSC.
- Z. Ying, L. Zhao, C. Zhang, Y. Yu, T. Liu, H. Cheng and F. Zhao, RSC Adv., 2015, 5, 42095–42100 RSC.
- J. Shang, S. Liu, X. Ma, L. Lu and Y. Deng, Green Chem., 2012, 14, 2899–2906 RSC.
- Z. Ying, Y. Dong, J. Wang, Y. Yu, Y. Zhou, Y. Sun, C. Zhang, H. Cheng and F. Zhao, Green Chem., 2016, 18, 2528–2533 RSC.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- H. P. SteinrÜk and P. Wasserscheid, Catal. Lett., 2015, 145, 380–397 CrossRef.
- Y. Gu and G. Li, Adv. Synth. Catal., 2009, 351, 817–847 CrossRef CAS.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS PubMed.
- K. Fukumoto, Y. Kohno and H. Ohno, Chem. Lett., 2006, 35, 1252–1253 CrossRef CAS.
- H. Ogura, K. Takeda, R. Tokue and T. Kobayashi, Synthesis, 1978, 394–396 CrossRef CAS.
- C. F. Cooper and S. J. Falcone, Synth. Commun., 1995, 25, 2467–2474 CrossRef CAS.
- A. Ion, V. Parvulescu, P. Jacobs and D. D. Vos, Green Chem., 2007, 9, 158–161 RSC.
- B. Yang, F. Zhou, S. Liu, P. Wang, A. S. Alshammari and Y. Deng, RSC Adv., 2015, 5, 48908–48915 RSC.
- M. Yoshida, N. Hara and S. Okuyama, Chem. Commun., 2000, 2, 151–152 RSC.
- M. M. Coleman, M. Sobkowiak, G. J. Pehlert, P. C. Painter and T. Iqbal, Macromol. Chem. Phys., 1997, 198, 117–136 CrossRef CAS.
- J. Mattia and P. Painter, Macromolecules, 2007, 40, 1546–1554 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra07452a |
| ‡ Peixue Wang and Xiangyuan Ma are co-first authors. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.