
Graphene oxide coated Fe3O4@mSiO2 NPs for magnetic controlled bioimaging
Abstract
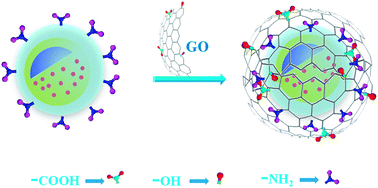
A novel GO coated porous magnetic NPs (Fe3O4@mSiO2@GO) for bioimaging was synthesized. First, Fe3O4 magnetic NPs were prepared through a solvothermal reaction. Second, through a surfactant-templating approach with cetyltrimethylammonium bromide (CTAB) as a template, a mesostructured CTAB/SiO2 composite was deposited on Fe3O4 NPs surfaces. Third, CTAB templates were removed to form a mesoporous SiO2 shell, resulting in mesoporous magnetic (Fe3O4@mSiO2) NPs. Forth, the magnetic NPs surfaces were functionalized with amino groups by (3-aminopropyl)triethoxysilane (APTES). Finally, the dye rhodamine B (RB) was loaded inside porous magnetic NPs, and the magnetic NPs surfaces were then wrapped with graphene oxide (GO) nanosheets. SEM analysis revealed that GO sheets have been successfully coated on magnetic NPs surfaces. The hysteresis loops analysis indicated that these magnetic NPs still retained strongly magnetic property. Furthermore, Fe3O4@mSiO2@GO NPs could efficiently protect the loaded dye from releasing. In addition, MTT assay revealed that the RB loaded Fe3O4@mSiO2@GO NPs exhibited insignificant cytotoxicity at moderate concentrations and the RB loaded Fe3O4@mSiO2@GO NPs could undergo target-directed move under the action of a magnetic field. The noncytotoxic magnetic hybrids presented significant potential for applications in cell imaging.
 Please wait while we load your content...
Please wait while we load your content...

