
Activation of peroxymonosulfate by iron-based catalysts for orange G degradation: role of hydroxylamine†
Abstract
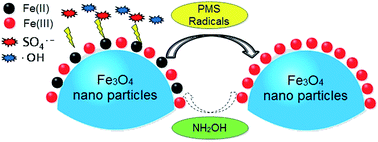
Magnetite nanoparticles (Fe3O4) have received considerable and increasing attention and been applied as activators for peroxymonosulfate (PMS) to generate reactive radicals for degrading organic contaminants. However, the slow transformation of Fe(III)/Fe(II) limits its repetitive and widespread application. Hence, hydroxylamine (NH2OH), a common reducing agent, was introduced to a Fe3O4/PMS system to accelerate the conversion of Fe(III)/Fe(II). With the addition of NH2OH, orange G (OG) degradation was largely increased at pH 2.0–7.0, and the generation of reactive radicals (sulfate- and hydroxyl-radicals) was significantly promoted. NH2OH was gradually degraded to N2, N2O, NO2−, and NO3−, while the eco-friendly gas, N2, was considered as its major end product. It was surprising to find that the catalytic ability of Fe3O4 soared gradually in the ten consecutive runs. The effectiveness of NH2OH for Fe3O4, Fe2O3 and iron sludge on activating PMS indicated its wide suitability for all the iron-based oxides/hydroxides. Continuous treatment of OG using 1.5 wt% Fe/Al2O3 catalyst (iron sludge immobilized on Al2O3) packed into a flow column reactor was performed, and no loss in activity over 30 d of operation was observed. Thus the present study provides a promising way to activate PMS for the rapid degradation of refractory organics and dispose the solid iron-based wastes.
 Please wait while we load your content...
Please wait while we load your content...

