
Cascade donor–acceptor organic ferroelectric layers, between graphene sheets, for solar cell applications
Małgorzata Wierzbowska*a and
Małgorzata Wawrzyniak-Adamczewskab
aInstitute of Physics, Polish Academy of Sciences (PAS), Al. Lotników 32/46, 02-668 Warszawa, Poland. E-mail: wierzbowska@ifpan.edu.pl
bFaculty of Physics, A. Mickiewicz University, ul. Umultowska 85, 61-614 Poznań, Poland
First published on 12th May 2016
Abstract
Organic ferroelectric layers sandwiched between graphene sheets are presented as a model of a solar cell. The investigated systems display many advantageous properties: (1) the cascade energy-level alignment; (2) simultaneous donor and acceptor character depending on the charge-carrier direction; (3) the charge-transfer excitonic type; (4) induced polarization of the electrodes, leading to a substantial work-function change of the anode and cathode – around ±1.5 eV.
I. Introduction
Power consumption and environmental pollution triggers an intensive search for efficient photovoltaic materials. One of the key factors for solar-cell efficiency is the suppressed recombination of charge carriers. This suppression can be achieved via introduction of charge trapping layers,1 and/or by the manipulation of energy-level alignment.2 Designing the heterostructure, which exhibits the cascade energy levels for the holes and electrons is tricky. On the other hand, if one applies multilayers of the same material – which, in addition, are ferroelectric – then the Stark effect shifts the energy levels of the subsequent layers gradually. Recently published theoretical studies by Sobolewski,3 of the ferroelectric columnar clusters in the context of the organic photovoltaics without p–n junctions, concern molecules with a dipole moment. The molecular orbitals of the subsequent energy levels in that work are well localized at the corresponding molecular rings in the stack; especially for the top and bottom molecules.Our systems are composed of flat molecules, namely 1,3,5-tricyano-2,4,6-tricarboxy-benzene. Its formula might be written as C6-3(NCCH2)-3(OCOH). These molecules consist of a central aromatic ring. Every second carbon of the ring is terminated with a cyano group possessing a dipole moment – standing out of the ring planes – and alternating with carboxy groups, which form the intermolecular hydrogen bonds within the planes. This is for the purpose of minimization of the electronic transport within the planes. The molecule with indexed atoms, as well as the top and side views of the single molecular layer are presented in Fig. 1. These networks can be stacked one on top of the other in various ways, but for the electronic transport the AA-type is favored. The considered structure resembles 2D covalent metal–organic frameworks,4,5 however intermolecular hydrogen bonds are used here instead of covalent bonds. These molecular layers are sandwiched between the graphene sheets that act as electrodes. The interlayer transport across our stacks is of the π–π type, and it strongly depends on the overlap between the pz orbitals of carbons within the molecular central part and those of the neighboring molecule, as well as the distance between the top atoms of the dipole group and the bottom hydrogens of the next top molecule.
 | ||
| Fig. 1 The building molecule with atomic indexes (left panel); top and side views of the hydrogen bonded hexagonal layer (right panel). | ||
Each ferroelectric monolayer in the stack acts as a donor of electrons to the next deeper layer, and simultaneously as an acceptor of electrons from the neighboring layer above. To our knowledge, this is the first system – except the columns in ref. 3 – where donor and acceptor (D–A) functionality is combined within the same molecule. Therefore, these layers will operate in a very similar way to the integrated heterojunctions5 or bulk heterojunctions,6,7 where two materials of different D–A functionality interpenetrate each other in the “bulk” of the optically active region.
Interestingly, the proximity of the molecular dipole moment polarizes the top and bottom electrode in opposite directions. The induced change of the surface dipole moment influences the work function, in such a way that it is higher for the anode and lower for the cathode. This is an advantageous feature. Similar properties are exhibited by graphene with the deposited ferroelectric layer (as shown in a recent patent8,9), with a documented efficiency of the photovoltaic effect of about 2%. The same idea of the addition of ferroelectric elements has been applied in 1D quantum-dot solar cells decorated with molecules possessing a dipole moment.10
II. Computational details
The density-functional theory calculations were performed using the QUANTUM ESPRESSO suite of codes (QE).11 This package is based on the plane-wave basis set and the pseudopotentials for the core electrons. The exchange-correlation functional was chosen for the gradient corrected Perdew–Burke–Ernzerhof parametrization.12 Ultrasoft pseudopotentials13 were used with energy cutoffs of 30 Ry and 300 Ry for the plane-waves and the density, respectively. It has been checked that increasing these values to 40 and 400 Ry, respectively, changes the band energies by less than 0.002 eV and the relative conformation energies of the ferroelectric and antiferroelectric molecular wire by less than 0.001 eV. The Monkhorst–Pack uniform k-mesh in the Brillouin zone has been set to 4 × 4 × 1 for the slab with two molecules per elementary cell, and 1 × 1 × 10 for the wire with one molecule per cell (for two molecules per cell, the corresponding k-mesh was 1 × 1 × 5), which was sufficient due to the large supercells. The vacuum separation between the periodic slabs was around 40 Å.In order to obtain the band structures projected onto the local groups of atoms, we employed the Wannier90 package,14 which interpolates bands using the maximally-localized Wannier functions.15,16 The same tool has been used for the calculation of the dipole moment and polarization. For the Wannier optimization procedure, we used the k-point mesh of the same accuracy as for the self-consistent DFT calculations. The number of Wannier functions was the same as the number of electrons in the system. The outer and inner windows were chosen in such a way that they were identical and their border energies were placed within the energy gaps between the groups of the composite bands. For metallic systems, such as a molecule between the graphene sheets, calculation of the Wannier occupation numbers was necessary. We used the k-point dependent procedure, being an extension of that used in the van der Waals functionals,17 and previously tested by us for GaAs doped with Mn.18
The dipole moment can be easily obtained from the maximally-localized-Wannier center positions, rn, using the formula
 | (1) |
III. Results
The geometries of the investigated systems have been optimized, and the intermolecular distance in the AA-type stack is 5.2 Å. The distance between the top molecule and the top graphene is the same as that between the molecules, and on the bottom it is smaller, of order 4.6 Å. The lateral intermolecular distance between the molecules can be described by the OH⋯O bond, which is 1.75 Å.A. Dipole moment
Systems presented in Fig. 1 are built of molecules containing two types of groups with the dipole moment: (1) COOH and (2) CH2CN. The first group is in the plane perpendicular to the plane of the central carbon ring. The dipole moment of this group is oriented from O to OH. Due to the fact, that this group plays the role of a chemical connection between the molecules, some molecules have the OH part above the carbon-ring plane and the neighboring molecules have this group below the central part plane. Therefore, in total there is no dipole moment originating from the intermolecular bonds within the layer, as it is in other hydrogen-bonded molecular systems.21,22 In contrast, the second group plays the role of the polarization source, with the dipole moment oriented from N down to the central C-ring plane.The calculated dipole moment of an isolated molecule is −2.06 Debye. When the molecules are placed in the column, then the proximity of other dipoles induce a larger polarization. The induced dipole moment in the column of molecules is presented in Fig. 2. The molecules placed in the middle of the finite number stack are characterized by the larger induced polarization – about 10% – than the molecules at the top and bottom of the column.
 | ||
| Fig. 2 Dipole moments induced in the column of molecules depending on their number in the stack (black circles), compared to the same value for the isolated molecules (red squares). The dipole moments induced at each molecule, when there are six of them in the stack, are also given (inside the green ellipses). | ||
We emphasize that the total dipole moment from the multilayer – i.e. the monolayers which are in the middle of the stack – does not cancel as in bulk ferroelectrics. This is because the interactions between the layers are of the van der Waals type, and molecules do not form chemical bonds between the layers. Thus, the electronic polarization at molecular dipoles affects the neighboring layer very weakly.
B. Cascade energy-level alignment and combined donor–acceptor functionality
The energy gap in the band structure of a single molecular layer is about 3.66 eV. For the 6-layer thick slab, the single-layer energy gap decreases to around 3.4 eV. While the total energy gap of the system closes due to the Stark effect, which shifts the top and bottom layer towards higher and lower energies, respectively. The intermediate layers shift gradually in energy, forming a cascade of the hole- and electron-energy levels, when moving step-by-step from the top to the bottom electrode. In Fig. 3, the density of states (DOS) projected onto each layer separately is presented, for the 2-, 4- and 6-layer stack. The overall energy gap for the 2-layer case is half of that for the single layer. In the case of the 4-layer stack, the total band gap closes. An increase of the number of molecular layers lowers the step of the energy shift of the neighboring layers. The shape of the projected DOS is only slightly changed between the layers and as a function of their number. In our case, a cascade energy-level alignment is obtained practically without any need for band engineering; which is the usual case when many materials are used in heterojunctions.2 Such a cascade is a very desirable property in order to avoid recombination of the charge carriers. | ||
| Fig. 3 Stark shift of the energy levels visible in the DOS projected at the layers; for 2-layer, 4-layer and 6-layer cases. | ||
The donor or acceptor character of each layer depends on its position with respect to the next layer – whether the carriers arrive from the anode or cathode – although all layers have an identical atomic structure. Due to our knowledge, this is the first material which shows the property of donor- and acceptor-type mimetism within the same layer. It is advantageous, because one can obtain the functionality of the sophisticated bulk heterojunctions6 by using only one 2D material stacked in multilayers.
Fig. 4 shows schematically the operation of the cascade energy-level system with the electrodes, whose bands energetics follow the sequence of the electron and hole energy levels of the optically active material. Each single optical excitation creates one hole–electron pair at one layer. Then, the holes move towards higher energies within the manifold of the valence band states and the electrons move down within the conduction band levels. In this way, the charge carrier transport across the optically active layers towards the electrodes takes place. Its efficiency is additionally factorized by the excitonic character and the work function of the molecular layers and the electrodes.
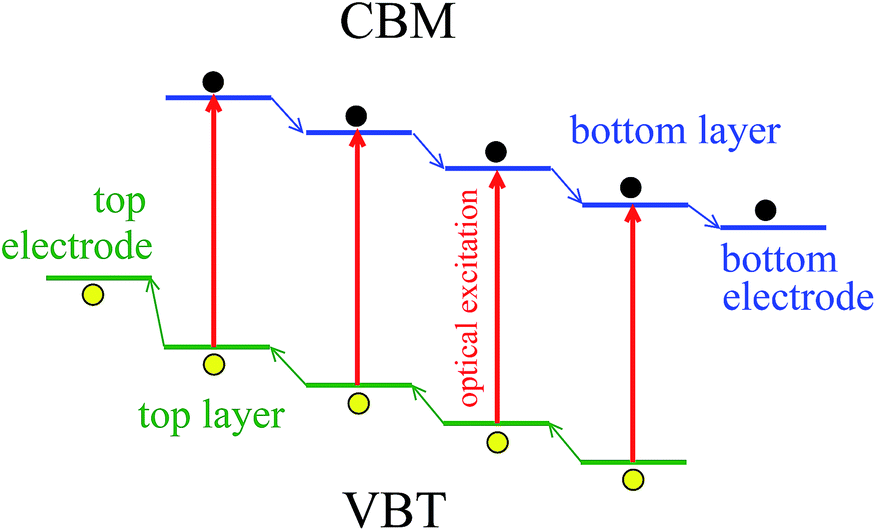 | ||
| Fig. 4 Scheme of the photovoltaic mechanism in the cascade energy-level system with the electrodes. VBT denotes the valence band top and CBM indicates the conduction band minimum. | ||
C. Exciton localization
Excitons – the electron–hole pairs – can be described as the difference of the charge distribution before and after the absorption of light. If this difference is spatially localized, we have so-called Frenkel excitons. The extended excitations are of the Wannier–Mott type. The excitons move through the system as a pair before splitting – which might end with the delayed fluorescence and recombination instead of the desired charge separation. Moreover, in molecular or very small quantum dot systems, the excitations lead to the singlet state, with a short life-time, or – after irradiative transition – to the longer lived triplet state. The excitonic localization character is one of the indicators of photovoltaic efficiency.Therefore, we analyze the atomic composition of the DOS at the hole and electron peaks in the VBT and CBM, respectively. In Fig. 5, we denote the central-ring carbons as C1, the adjacent carbons as C2, and the further carbons connected to N as C3. The DOS projected at these groups of carbons, as well as nitrogens and oxygens, is pictured for the peak maximum at the VBT and CBM; separately for each molecular plane of the 4-layer system. The values are normalized to the total DOS and given in percentage. It is important to note that the lowest excited electronic states are composed mainly of carbons in the central ring. The hole states are delocalized over the dipole groups. The component from the central part of the molecule to the hole states is only ∼10%. The above observations are true for all layers in the stack. As follows, our electron–hole pairs result from the excitation of the valence electrons localized around the anion (groups containing N) into empty states localized around cation (the central benzene ring). Such pairs are called charge-transfer excitons. A quick estimation for the Bohr exciton radius in our systems is about 4.7 Å. It is defined as the distance between the center of the molecule and one of the nitrogen atoms. Therefore, we have Frenkel-type charge-transfer excitonic material. Interestingly, the charge-transfer excitons are also formed at the van der Waals interfaces of other materials.23
 | ||
| Fig. 5 The DOS projected at atoms indexed in Fig. 1. The presented values were taken at maxima of the VBT and CBM peaks, of the 4-layer system shown in Fig. 3. The numbers are given as a percentage of the total DOS. | ||
For further insight into the localization of electrons and holes, we project the band structures onto those maximally-localized Wannier functions which are centred near chosen groups of atoms. These plots are presented in Fig. 6 for carbons of the central ring, nitrogens and oxygens. The Figure confirms our previous result, that the central-ring atoms are involved in the conduction states and the dipole and oxygen groups in the valence states. Moreover, we can see that using the OH⋯O bonds prohibits the electronic and hole transport within the molecular planes; which is obvious from the flat band structures.
 | ||
| Fig. 6 Band structure of the single molecular layer projected at the carbons of the central ring (C1) and nitrogens and oxygens, respectively. The color scale indicates the factor of the near-atom-centred Wannier functions in band decomposition. | ||
D. Work function change caused by proximity of ferroelectric layers
The dipole moment of molecules induces charge polarization within the graphene layers. In order to estimate the strength of this polarization, we performed calculations for the periodic system with a large elementary cell, which contains a single molecule between the graphene layers on the top and bottom. The number of carbon atoms in each graphene part in that cell is 160. The top and side views on this elementary cell are presented in Fig. 7. The Figure also contains the plot of the DOS projected onto the molecule and the graphene sheets. The DOS shift of the top and bottom graphene layers, by about ±0.2 eV for each electrode, is caused by the Stark effect and leads to a small charge transfer between the leads. | ||
| Fig. 7 Top and side views of a single molecule between the graphene sheets and the projected DOS. | ||
Using the Wannier-functions technique, described in the “Computational details”, we estimated the induced dipole moment in the top graphene as −2.58 Debye and the bottom graphene as +2.54 Debye; within the elementary cell with each graphene surface around 411 Å2. The above result is promising for tuning the work function, since the opposite electrodes polarize differently. It is convenient, for the power of the photovoltaic conversion, to chose the anode electrode as a high work function material and the cathode as a low work function metal.
The experimental value of the work function of the pristine graphene is around 4.6 eV.24 It is possible to tune this property with the electric field.25 The same effect can be achieved by a decoration of the graphene surface with metals,26 or any 2D system with the molecules with a dipole moment.27,28
The change of the work function ΔΦ caused by the induced polarization ΔP is given by the Wigner and Bardeen formula29,30
 | (2) |
Induced polarizations obtained for the top and bottom electrodes cause the change of the work function by 1.53 eV for the anode and −1.50 eV for the cathode. The surface of the presented “square” elementary cell is smaller than that of the molecular layer in the hexagonal lattice in Fig. 1 – by a factor of 0.9066 per molecule. This is due to the large “hole” within the molecular hexagons. Therefore, we estimate that the average change of the work function of the graphene leads, applied to the layers proposed in Fig. 1, is around 1.39 eV and −1.36 eV for the anode and cathode, respectively. Since the polarization induced in graphene near so much porous molecular lattice is not homogeneous, one can expect that the “effective” local change of the work function is much larger. The work function changes of the polarized graphene electrodes are not so large when one compares them with these changes of about 2 eV in the two-dimensional transition metal carbides and nitrides functionalized with OH.27
E. Antiferroelectric arrangement, formation energies with graphene
During the growth of the molecular crystal, it might occur that some rings adsorb on each other with opposite sides. In such case, locally, the antiferroelectric phase will form. To estimate the probability of such event, we calculated the 1D molecular wires oriented in: (i) the ferroelectric; (ii) the totally antiferroelectric (with total dipole moment 0); and (iii) the not completely antiferroelectric manner (with the total dipole moment lowered with respect to the ferroelectric case but not equal to 0). The two antiferroelectric cases are presented in Fig. 8. They differ from each other by a flip of H in the COOH groups. The orientation of the benzene centers in the ferroelectric case (FE) is of the AA-stacking type (one on top of the other). In the antiferroelectric cases (AFE), we rotated one molecule with respect to the other by 30 degrees. This rotation does not change the dipole moment along the molecular wire. There is strong electrostatic repulsion within the planes rich in H atoms and the planes rich in N and O atoms. For the first geometric conformation, the optimized distances between the central C-rings are 5.51 and 4.89 Å for the N-rich and H-rich planes, respectively. The corresponding numbers for the second geometric conformation are 5.55 and 4.85 Å, respectively. In both cases, the total energy per molecule of the ferroelectric case is lower than this energy of the antiferroelectric case: (i) for the first conformation with vanishing dipole moment, the FE-AFE energy difference is −675 meV; and (ii) for the second conformation with a small dipole moment, the corresponding energy difference is −302 meV. | ||
| Fig. 8 The top (left panel) and side (middle and right panel) view of the two conformations of the antiferroelectric wire. | ||
The formation energy Ef, is defined as the difference of the total energies of the whole system and the two subsystems,
| Ef = EA+B − EA − EB. | (3) |
For a single molecule on graphene, the formation energy is 1.33 eV for the bottom graphene layer (below the H-rich side of the molecule) and 1.34 eV for the top graphene layer (above the N-rich side of the molecule). The formation of the ferroelectric wire is exothermic with 622 meV per molecule, while for the totally antiferroelectric wire, it is endothermic with 53 meV per molecule. Therefore, the adsorption of the second and subsequent layers on top of graphene will be easier than that of the first layer.
Detailed geometries of the molecule in vacuum, in the wire and on graphene are given in Table 1.
| M | M@g | g@M | FE | AFE | |
|---|---|---|---|---|---|
| Bond lengths [Å] | |||||
| C-ring | 1.405 | 1.434 | 1.434 | 1.405 | 1.412 |
| C-ring–C(OOH) | 1.504 | 1.545 | 1.544 | 1.511 | 1.508 |
| C-ring–C(CN) | 1.527 | 1.525 | 1.524 | 1.523 | 1.528 |
| C–C(N) | 1.468 | 1.422 | 1.421 | 1.460 | 1.465 |
| C–N | 1.163 | 1.269 | 1.268 | 1.165 | 1.165 |
| C–O(H) | 1.370 | 1.389 | 1.390 | 1.358 | 1.376 |
| C–O | 1.215 | 1.171 | 1.170 | 1.218 | 1.209 |
| O–H | 0.984 | 1.004 | 1.004 | 0.986 | 0.987 |
| C–H | 1.102 | 1.151 | 1.150 | 1.100 | 1.104 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Angles [deg] | |||||
| C–C–C(N) | 116.7 | 113.2 | 113.2 | 114.1 | 118.0 |
| C–C–N | 175.9 | 166.1 | 166.2 | 178.4 | 173.3 |
| O–C–O(H) | 123.3 | 128.2 | 128.2 | 124.8 | 122.8 |
IV. Conclusions
We have shown that the organic ferroelectric layers with the lateral OH⋯O bonds display a variety of desired properties for photovoltaic efficiency. When these layers are AA-type stacked, they are similar to columnar clusters with a dipole moment, which were recently proposed as photovoltaic systems without the p–n junction.3 Ferroelectricity causes the cascade energy-level alignment of the hole and electron states of the subsequent molecular layers. Importantly, the dipole moments of the layers from the middle of the stack do not cancel, as in the case of bulk ferroelectrics where only the surface dipole moments resist. The fabrication of the presented layers could be conducted in the presence of an electric field, such that the ferroelectric (FE) and not the antiferroelectric (AFE) alignment of the neighboring layers is achieved. Statistically, mixed FE and AFE ordering occurs. However, the AFE orientation is energetically less favourable than the FE one by about 0.30–0.68 eV per molecule, depending on the partial or total vanishing of the dipole moment originating from the COOH and CH2CN groups.Moreover, the donor or acceptor character of each layer depends on the direction from which the charge carrier arrives, since this chemical property is defined with respect to the neighboring layers. The optically generated hole–electron pair has the character of a charge-transfer exciton, with an estimated Bohr radius of about 4.7 Å. The work function of the graphene electrodes, applied to such system, changes due to the induced surface polarization – and it is about ±1.4 eV for the anode and cathode leads, respectively, when a monolayer of molecules is sandwiched. The functionality of the presented system can be compared to that of the integrated heterojunctions.5 The addition of the electron and hole transport layers is not necessary. Especially, that these systems also possess the property of separate paths for electron and hole transport across the layers.31
Acknowledgements
This work has been supported by The National Science Centre of Poland (the Projects No. 2013/11/B/ST3/04041 and DEC-2012/07/B/ST3/03412). Calculations have been performed in the Cyfronet Computer Centre, using the Prometheus computer which is a part of the PL-Grid Infrastructure, and by part in the Interdisciplinary Centre of Mathematical and Computer Modeling (ICM).References
- J. Liu, G. H. Kim, Y. Xue, J. Y. Kim, J. B. Baek, M. Durstock and L. Dai, Adv. Mater., 2014, 26, 786 CrossRef CAS PubMed
.
- Z.-K. Tan, K. Johnson, Y. Vaynzof, A. A. Bakulin, L.-L. Chua, P. K. H. Ho and R. H. Friend, Adv. Mater., 2013, 25, 4131 CrossRef CAS PubMed
- A. L. Sobolewski, Phys. Chem. Chem. Phys., 2015, 17, 20580 RSC
- D. D. Medina, J. M. Rotter, Y. Hu, M. Dogru, V. Werner, F. Auras, J. T. Markiewicz, P. Knochel and T. Bein, J. Am. Chem. Soc., 2015, 137, 1016 CrossRef CAS PubMed
- M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802 CrossRef CAS PubMed
- Z. Xiao, Y. Yuan, B. Yang, Y. VanDerslice, J. Chen, O. Dyck, G. Duscher and J. Huang, Adv. Mater., 2014, 26, 3068 CrossRef CAS PubMed
- S. Lattante, Electronics, 2014, 3, 132 CrossRef CAS
- B. Ozyilmaz, et al., WO Patent App. PCT/SG2013/000, 2013, p. 114
- K. Kim, S.-H. Bae, C. T. Toh, H. Kim, J. H. Cho, D. Whang, T.-W. Lee, B. Özyilmaz and J.-H. Ahn, ACS Appl. Mater. Interfaces, 2014, 6, 3299 CAS
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulović, ACS Nano, 2014, 8, 5863 CrossRef CAS PubMed
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car and C. Cavvazzoni, et al., J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- D. Vanderbilt, Phys. Rev. B: Condens. Matter, 1990, 41, R7892 CrossRef
- A. A. Mostofi, J. R. Yates, Y. S. Lee, I. Souza, D. Vanderbilt and N. Marzari, Comput. Phys. Commun., 2008, 178, 685 CrossRef CAS
- N. Marzari and D. Vanderbilt, Phys. Rev. B: Condens. Matter, 1997, 56, 12847 CrossRef CAS
- N. Marzari, A. A. Mostofi, J. R. Yates, I. Souza and D. Vanderbilt, Rev. Mod. Phys., 2012, 84, 1419 CrossRef CAS
- L. Andrinopoulos, N. D. M. Hine and A. A. Mostofi, J. Chem. Phys., 2011, 135, 154105 CrossRef PubMed
- K. Z. Milowska and M. Wierzbowska, Chem. Phys., 2014, 430, 7 CrossRef CAS
- R. Resta, Rev. Mod. Phys., 1994, 66, 899 CrossRef CAS
- M. Gibertini, G. Pizzi and N. Marzari, Nat. Commun., 2014, 5, 5157 CrossRef CAS PubMed
- S. Horiuchi, R. Kumai and Y. Tokura, J. Am. Chem. Soc., 2013, 135, 4492 CrossRef CAS PubMed
- S. Horiuchi, K. Kobayashi, R. Kumai and S. Ishibashi, Chem. Lett., 2014, 43, 26 CrossRef CAS
- X. Zhu, N. R. Monahan, Z. Gong, H. Zhu, K. W. Williams and C. A. Nelson, J. Am. Chem. Soc., 2015, 137, 8313 CrossRef CAS PubMed
- S. Suzuki, C. Bower, Y. Watanabe and O. Zhou, Appl. Phys. Lett., 2000, 76, 4007 CrossRef CAS
- Y.-J. Yu, Y. Zhao, S. Ryu, L. E. Brus, K. S. Kim and P. Kim, Nano Lett., 2009, 9, 3430 CrossRef CAS PubMed
- K. T. Chan, J. B. Neaton and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235430 CrossRef
- M. Khazaei, M. Arai, T. Sasaki, A. Ranjbar, Y. Liang and S. Yunoki, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 075411 CrossRef
- W. E. Ford, D. Gao, N. Knorr, R. Wirtz, F. Scholz, Z. Karipidou, K. Ogasawara, S. Rosselli, V. Rodin, G. Nelles and F. von Wrochem, ACS Nano, 2014, 8, 9173 CrossRef CAS PubMed
- E. Wigner and J. Bardeen, Phys. Rev., 1935, 48, 84 CrossRef CAS
- T. C. Leung, C. L. Kao, W. S. Su, Y. J. Feng and C. T. Chan, Phys. Rev. B: Condens. Matter, 2003, 68, 195408 CrossRef
- M. Wawrzyniak-Adamczewska and M. Wierzbowska, J. Phys. Chem. C, 2016, 120, 7748 CAS
| This journal is © The Royal Society of Chemistry 2016 |