
Nickel-catalyzed α-benzylation of sulfones with esters via C–O activation†
Abstract
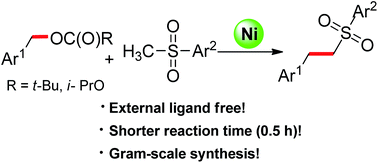
The nickel-catalyzed α-benzylation of sulfones with readily available benzylic alcohol derivatives was achieved via C–O activation. The transformation was complete in 30 minutes using a simple Ni(COD)2 as a catalyst without any additional ligands. A variety of substituted sulfones including the building block for bioactive compound eletriptan were synthesized by using the strategy.
 Please wait while we load your content...
Please wait while we load your content...

