
Dendrimeric based microbicides against sexual transmitted infections associated to heparan sulfate
Rafael Ceña-Díezabc,
Daniel Sepúlveda-Crespoabc,
Marek Malyde and
Mª Angeles Muñoz-Fernández*abc
aLaboratorio InmunoBiología Molecular, Hospital General Universitario Gregorio Marañón, C/Dr Esquerdo 46, 28007 Madrid, Spain. E-mail: mmunoz.hgugm@gmail.com; Tel: +34 915 868 565
bInstituto de Investigación Sanitaria Gregorio Marañón (IiSGM), Spanish HIV-HGM BioBank, Madrid, Spain
cNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Madrid, Spain
dDepartment of Innovative Technologies, University of Applied Science of Southern Switzerland, Switzerland
eFaculty of Science, J. E. Purkinje University, Usti n. L., Czech Republic
First published on 20th April 2016
Abstract
Cell surface heparan sulfate (HS) represents a common link that many sexually transmitted infections (STIs) require for infection. The role of HS is associated with several viral STIs, which include those caused by herpes simplex virus (HVS), human immunodeficiency virus (HIV), human papillomavirus (HPV) and hepatitis C virus (HCV). Nowadays, no cure has been found for any of the STIs associated with viral pathogens. In this review, we evaluate dendrimers such as peptide derivatized-dendrimers, carbosilane dendrimers, polysulfated galactose functionalized glycodendrimers and PAMAM dendrimers, among others, as potential candidates for the development of topical microbicides against viral STIs associated with HS. Subsequently, we propose the mechanism of action of these dendrimers and how it might be associated with the relevance of HS in several STIs. HS in just an ancillary factor in the case of HIV-1 infection and it plays a major role in the case of HCV, HSV-2 and HPV viruses. However, the mechanism of action presented by these dendrimers is common in all pathologies, acting at the level of viral entry into the target cell, either directly blocking the viral particles that are meant to bind to the HS or binding to cellular co-receptors.
Introduction
Sexually transmitted infections (STIs) are a major global cause of acute diseases, infertility, mobility and death with serious medical and psychological consequences for millions of men, women and infants. There are over 30 bacterial, viral and parasitic pathogens that have been identified to date that can be transmitted sexually (Table 1). In recent years, sexually transmitted diseases (STDs) have become a major economic as well as public health problem. In 2008, there were an estimated 110 million prevalent STIs and around 19.7 million new cases just in the United States.1,2| Pathogen | GAG ligand | Reference |
|---|---|---|
| a GAG, glycosaminoglycan; HS, heparan sulfate; VLP, virus like particles; Omp, outer membrane protein; OpA, opacity protein; DsrA, H. ducreyi serum resistance antigen A; SGSP1, submandibular gland secreted proteoglycan 1. | ||
| Viruses | ||
| HSV-1 | HS gC/D | 34,99 and 100 |
| HSV-2 | HS gB/C | 101 |
| VZV | HS gB/C | 33 and 34 |
| Human cytomegalovirus | HS gB | 102 |
| HPV | HS–VLP | 103 |
| HIV-1 & HIV-2 | HS–gp120; gp41 | 67 and 104 |
| HBV & HCV | HS–L-viral envelope protein | 88 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Bacteria | ||
| Chlamydia trachomatis | HS–Omp | 105 |
| Treponema pallidum | HS–fibronectin | 106 |
| Neisseria gonorrhoea | HS–OpA | 107 |
| Haemophilus ducreyi | DsrA–fibronectin/HS | 108 |
|
||
| Fungi | ||
| Candida albicans | HS–SGSP1/ECM-proteins | 109 |
In 2012 World Health Organization published an estimation of the incidence and prevalence of four curable STIs – Chlamydia trachomatis, Neisseria gonorrhoeae, syphilis and Trichomonas vaginalis – in adults between 15 and 49 years of age. The estimated number of cases in 2008 was to be close to 500 million: 105.7 million cases of Chlamydia trachomatis, 106.1 million cases of Neisseria gonorrhoeae, 10.6 million cases of syphilis and 276.4 million cases of Trichomonas vaginalis.
Among all the pathogens responsible for different STIs, viruses play a major role. It is estimated that several hundred million individuals worldwide are infected with viral STDs including herpes simplex virus type 2 (HSV-2), human immunodeficiency virus (HIV) and human papillomaviruses (HPV). However, no cure has been found for any of the STIs associated with viral pathogens, whereas sexual diseases associated with bacterial or parasitic pathogens with higher worldwide prevalence have a functional cure. The need to deal with these viral infections becomes more obvious, not only because they represent a problem for public health, but also a serious economic problem. Costs associated with HIV infection ($12.6 [$9.5–$15.7] billion) in United States accounted for more than 81% of the total lifetime direct medical cost of all STIs in 2008.1 Despite all the recent efforts and progress neither cure has been found nor success in slowing the progression of these diseases has been achieved.3 Although the emphasis has been on preventing HIV, protection from other STIs is desirable in terms of avoiding diseases and associated consequences. Importantly, many STIs may increase vulnerability to HIV infection (e.g., genital herpes infection facilitates the transmission of HIV).4
Any common element which enables to link the main STIs represents an ideal target for the development of new topical microbicides that would curb the transmission of different STIs. Such a link would be an immense therapeutic or prophylactic value.
Heparan sulfate proteoglycans (HSPGs) are glycoproteins, with the common characteristic of containing one or more covalently attached heparan sulfate (HS) chains, a type of glycosaminoglycan (GAG) (Fig. 1).5 HS are linear sulfated polysaccharides that through covalent links bind to HSPGs core proteins on cell surfaces and in extracellular matrixes. HS is ubiquitously expressed on the cell surface and in the extracellular matrix of almost all cell types as HSPGs.6 Undesirable interactions between HSPGs and their protein partners may result in the development of certain pathologies, such as chronic inflammation, angiogenesis, tumor growth or microbial infections.5 In addition, most human viruses bind to cellular glycans in order to carry out their critical initial entry step. The primary attachment requires a low affinity interaction between basic binding pockets of the virion glycoproteins and negatively charged HS moieties in cellular GAGs.7
 | ||
| Fig. 1 Structural scheme of heparan sulfate. HS are linear polymers composed of repeating uronic acid [D-glucuronic acid (GlcA) or L-iduronic acid (IdoA)] and D-glucosamine (GlcN) disaccharide subunits. HS biosynthesis is started by the attachment of xylose to specific residues in HSPG core proteins, followed by the formation of a linking tetrasaccharide (xylose–galactose–galactose–glucuronic acid). The arrow shows the 3-O position of the glucosamine residue where sulfation is essential for HSV-1 and HSV-2 glycoprotein D (gD) binding. | ||
In this review we highlight new dendrimeric treatments for mitigating the increasing prevalence of viral STIs associated with HS.
Role of heparan sulfate in dendrimers against HSV-2 infection
Currently, there is neither cure for HSV-2 persistent infection nor effective vaccines. HSV-2 pathology can be mitigated or modulated through current antiviral treatment, but latent infection cannot be eliminated.8–11 Prevention strategies are essential to fight against HSV-2. The presence of a genital herpetic infection may increase an individual's chances of becoming infected with HIV-1 3-fold to 4-fold.4 Preventing HSV-2 infection would also indirectly decrease the number of HIV-1 new infections in a significant manner.The initial step in mucosal infection by the HSV-2 requires its binding to certain GAGs naturally present on host cell membranes. HS and 3-O sulfated HS (3-OS HS) have a major clinical significance during HSV-2 entry to the host cell. 3-OS HS is produced after an enzymatic modification of HS catalyzed by 3-O-sulfotransferases (3-OSTs).6,12
There is almost no variability close to gB sequence (98–99% identity) or between HSV-1 and HSV-2 sequences (85% identity). This very low variability is observed in their lysine-rich region. Therefore, it has been speculated that this variability could imply recognition of different structural features in HS in each form of gB.
The current model of HSV-2 entry, five glycoproteins (gB, gC, gD and the heterodimer gH/gL) play an important role in the HSV-2 fusion between the viral envelope and the membrane of the host cells. The initial attachment or binding step requires viral gB binding to unmodified HS,13,14 starting a phenomenon for the transport of extracellular virus particles, termed “viral surfing”, which has already been seen in other viruses, such as retroviruses and HPV type-16.15,16 In the next step, the process is triggered by conformational changes in viral gD, specifically recognizes 3-OS HS, among other receptors, and this interaction facilitates an hemifusion state. Alteration in gD conformation then mobilizes the heterodimer gH and gL and gB to initiate the fusion process at the cell surface.13,14 Due to the complex fusion mechanism of HSV-2, which requires four viral glycoproteins, and because there are only structural data for gB and gD, many questions on the mechanism by which HSV-2 fuses its envelope with the host cell membrane remain unresolved (Fig. 2).
 | ||
| Fig. 2 Schematic representation of heparan sulfate and 3-O sulfated heparan sulfate during HSV-2 entry to the host cell. (A) Initial virus attachment to unmodified HS, which is mediated by HSV-2 glycoprotein B (gB), followed by a process of surfing which increase viral attachment strength. (B) This interaction leads to HSV-2 glycoprotein D (gD) to interact with at least one of the following co-receptors: the modified form of HS (3-OS HS), herpes virus entry mediator (HVEM) or nectin-1 or 2, which promotes virus–cell membrane hemifusion state. (C) Alteration in gD conformation then mobilizes the heterodimer gH and gL and gB to initiate the fusion process at the cell surface and (D) release of viral genomic material. | ||
In the case of the HSV-2, compounds targeting steps of the viral cycle linked to HS would offer a high efficiency prophylactic strategy due to the great importance of this receptor in the entry mechanism of HSV-2 to the host cell. Several dendrimers offer such a strategy.
A screening of 15 peptide derivatized-dendrimers with differences in the amino acid sequences of their surface groups led to the selection of SB105 and SB105_A10. SB105 and SB105_A10 have a potent dose-dependent antiviral activity against HSV-2, the mechanism of action involves the inhibition of the virion attachment to HSPGs on the surface of target cells.17 SB105 and SB105_A10 are peptide-derivatized dendrimers developed from the M6 prototype.18 The anti-HSV activity of SB105 and SB105_A10 depends on the specific amino acid sequence of the peptide surface groups (QKKIRVRLSA) attached to the dendrimer, as variations of this sequence were found to reduce or abolish their antiviral action. It is likely that their action mechanism could be due to ionic interactions between their positively charged amino acids and the negatively charged sulfate and carboxyl groups in HS. As it occurs with HIV, SB105 and SB105_A10 bind to the negatively charged sulfate and carboxyl groups of HS chains, blocking the binding of HSV-2 to cellular receptors. Later it was shown that SB105_A10 binds to cells in a HS-dependent manner, since this interaction was prevented by heparinase/heparitinase treatment of target cells.19 Therefore, SB105 and SB105_A10 neither form complexes with HSV envelope glycoproteins nor inhibit HSV-2 DNA synthesis.
Several dendrimers that had shown their ability against HIV, three PAMAM (polyamidoamine) and two polylysine dendrimers (SPL-2999 and SPL-6741), were evaluated in vitro for antiviral activity against HSV-2.20 All compounds showed in vitro antiviral activity against HSV-2. When they were added to target cells prior to the addition of the virus, they completely halted both viral adsorption and penetration into susceptible cells. However, only SPL-2999 was able to inhibit DNA synthesis in HSV-2 infected cells. SPL299 appears to have secondary mechanisms of action, in addition to the postulated dendrimers' primary antiviral mechanism, which occurs early in the infection process, possibly blocking virus attachment to the cell or interfering with adsorption.20–22 It has been postulated that the ability of these dendrimers to disrupt HSV-2 initial binding through viral gB recognition of HS and 3-OS HS of the host cell might be due to their electrostatic nature and functionalized groups in their capping layer, which are naphthyl disodium disulfonate for SPL-2999 and phenyl disodium dicarboxylate for SPL6741.20–22
Then the earlier thiourea linkage of SLP-2923, SLP-2784, SLP-2999 and SLP-6039 was replaced by a more stable amide bond identifying three new microbicide candidates, SPL7013, SPL7015 and SPL7032, which are all prepared of the same lysine. These three microbicide candidates had similar in vitro activities against HSV-2.22
The mechanism of HSV-2 inhibitory action, evaluated by time-of-addition studies, is postulated to interfere both the cell entry, likely via HS, and HSV-2 replication. This suggests that the microbicide candidates may have high potential as a therapeutic agent against HSV-2 infections.22–24 The late stage of HSV-2 inhibitory mechanism is probably associated with interference in DNA synthesis, and this is the way how the SLP-2999 works. SPL7013 showed strong anti-HSV-2 activity by inhibiting virus internalization and blocking HSV-2 adsorption to host cells.24 It is highly possible that SPL7013 dendrimer interferes in the initial binding of HSV-2 with gB or gD of HS.25 This inhibition might be due to the interaction between their positively charged amino acids and the negatively charged sulfate and carboxyl groups in HS.23,24 One potential handicap of the polyanions in development as topical microbicides is that they are mixtures of compounds. However, SPL7013 polylysine dendrimer has been characterized by mass spectrometry, capillary electrophoresis, and high-pressure liquid chromatography, and in-process controls SPL7013 has been developed to tightly control the synthesis. As a result SPL7013 was the only one to reach preclinical trial as a topical microbicide.
HSV membrane fusion represents an attractive target for anti-HSV therapy. It has been reported that fusion peptides when added exogenously inhibit viral fusion, probably by intercalating with viral fusion proteins upon adopting functional structure in membranes.26 Due to previous studies that showed that peptides derived from certain regions of gH have the potential to interact with membranes,26–28 a set of peptides containing mutations in one of these domains, gH-(625–644) were synthesized.27 Poly(amide)-based dendrimer functionalized at the termini with a membrane-interacting peptide derived gH625 from the HSV-1/HSV-2 was selected by its ability to interact with model membranes and to inhibit viral infectivity.29 The gH625 peptide acts by binding to gH through oligomerization of the gH625–644 domain present on the gH, in a probable rearrangement of its structural domains, preventing a complete and functional interaction between gH and the membrane to fuse. Alternatively, the gH sequence presents on the peptidodendrimer may interact with other glycoproteins present on the virion envelope, such as gB or gD. This approach provides several advantageous features, such as target specificity, low toxicity, and the possibility to modify surface characteristics easily.29
Recently, a screening of eight polyanionic carbosilane dendrimers with anti-HIV activity led to the selection of G1-S4, G2-S16 and G3-S16 as candidates with strong antiviral activity against HSV-2.30 The results suggested that the first antiviral mode of action of dendrimers occurs early in the infection process, likely blocking HSV-2 binding and internalization. However, G2-S16 against HSV-2 had a second mechanism. G1-S4 and G3-S16 have sulfate end-groups and bind directly on viral proteins on the surface of HSV-2, inactivating them. These results are in consonance with previous studies with other sulfonated-terminated dendrimers.23,24 Molecular modeling of the binding of dendrimers with gB31,32 showed that the smaller and more flexible G1-S4 dendrimer blocked directly the virions hindering HSV-2 fusion through gB, reaching worse accessible binding sites of gB (Fig. 3).30 As in the case of SPL7013, G1-S4 and G2-S16 conduct their inhibitory effect against HSV-2 by halting HVS-2 interaction with HS. Whereas, G1-S4 sulphated dendrimer acts as analogue of HS and binds to the HSV-2 gB. G2-S16 dendrimer due to its big size is unable to reach inhibitory binding sites in the gB. However, G2-S16 binds to the proteins on the HSV-2 surface as this dendrimer does in the case of the HIV-1 infection.
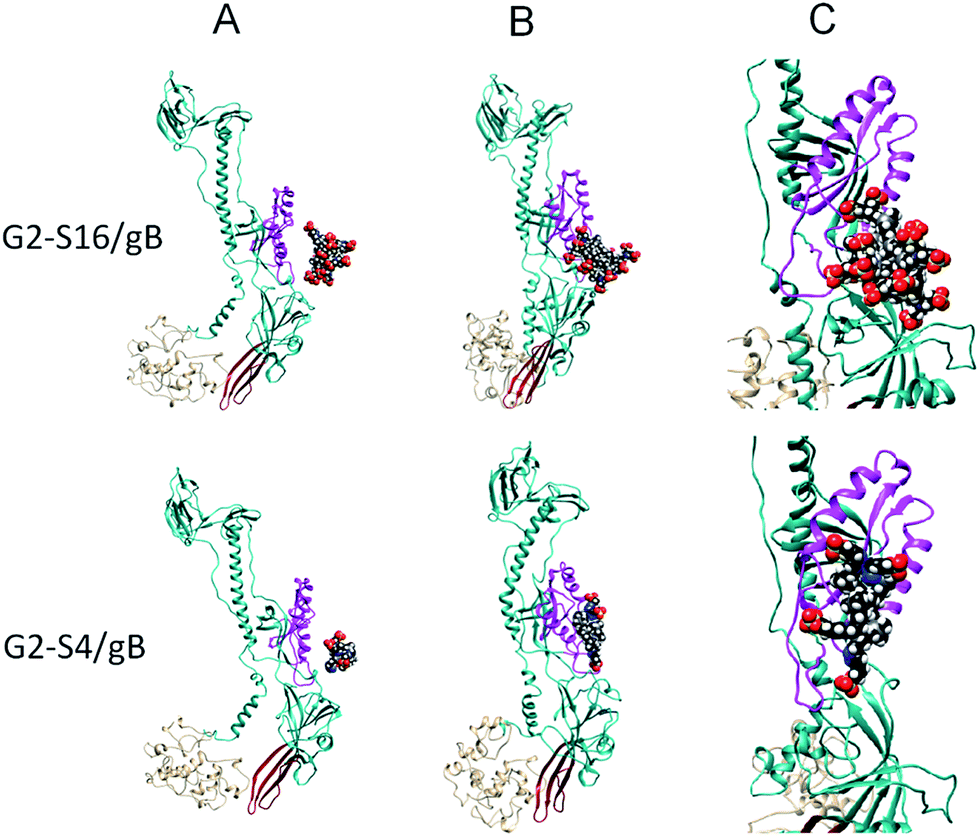 | ||
| Fig. 3 Molecular modeling of the interaction of dendrimers with gB at gH–gL binding area. (A) Initial configurations of simulated G2-S16/gB (top), G1-S4/gB (bottom) systems. (B) Final G2-S16/gB (top) and G1-S4/gB (bottom) complexes obtained from the initial configurations. (C) Detail view of the final G2-S16/gB (top), G1-S4/gB (bottom) systems. Notes: the main part of the suggested binding interface for the gH–gL is highlighted in magenta. The putative fusion loops are in red and the C-terminal part comprising residues 721–904 is in tan. Atoms of dendrimers are coloured as follows: C, black; O, red; Si, gray; S, yellow; N, blue; H, white. | ||
Due to the capacity of these dendrimers to inhibit HSV-2 infection through blocking of HS interaction with viral gB or gH–gL/gB and hence blocking the proper initiation of the fusion machinery, it would be of great interest to evaluate the protective ability of these dendrimers against other member of the herpes virus family, which are known to infect humans. These other members are varicella-zoster virus (VZV), cytomegalovirus (CMV), Epstein–Barr virus (EBV), human herpes virus 6 (HHV-6), HHV-7, and HHV-8 or Kaposi sarcoma-associated herpes virus.
However, not all herpes viruses are dependent on HS for their attachment. HS has a major role as a receptor for VZV binding. The VZV gB has been demonstrated to bind heparin, and gB soluble binds to cells expressing HS but poorly to cells devoid of this GAG. Therefore, it is likely that HS has a role as a receptor for VZV binding.33,34 As in the case of VZV, the initial binding of human CMV with host cells is through interaction with HS.35,36
Role of heparan sulfate in dendrimers against HIV infection
HIV-1 replication cycle offers many potential targets for microbicide development.37 However, both the inhibition of entry and retrotranscription processes are currently considered the most promising strategies.38Two glycoproteins on the viral envelope, gp120 and gp41, are essential for the entry of the virus to the host cell. Protein gp120 binds to the CD4 receptor on the membrane of the target cell and then undergoes a conformational change, which causes the exposure of the V1/V2 and V3 loop.39,40 This shift produces the interaction between gp120 and the CXCR4 and CCR5 co-receptors and consequently the fusion of the gp41 ectodomain with the cell membrane.41 Four positively charged domains identified in the V2 and V3 loops, in the C-terminal domain and within the CD4-induced bridging sheet of the gp120 play an important role in enhancing the virus–cell interaction. These domains interact with the negatively charged HSPGs on the target cell membrane or with cell membrane lectin-binding proteins such as DC-SIGN.42–44
HS is considered an ancillary binding factor for HIV-1 in macrophages, dendritic cells and epithelial and endothelial cells.45–48 HSPGs play an important role in HIV adsorption and entry, HIV-1 exploits the anionic HS chains of HSPGs to attach and enter to the host cell.49 The ability of HS to recognize HIV-1 appears to be associated to the gp120, although it has been demonstrated that HS not only interacts with HIV-1 gp120, but also interacts with the fusion gp41 of HIV-1. The most interesting fact about HIV-1 attachment mediation via HS is associated with HIV-1 binding to low CD4 expression cells. HS may compensate this deficiency in CD4 favoring viral particle concentration at the cell surface and, therefore, increasing HIV binding which would lead to increased infectivity.50
Recently, peptide derivatized-dendrimer on a lysine core with the peptide sequence ASLRVRIKK (SB105-A10) exhibited antiviral activity against several HIV-1 isolates from HIV+ individuals in peripheral blood mononuclear cells, prevented the HIV attachment/entry by multiple mechanisms. In addition to the direct activity of SB105-A10 on the envelope glycoproteins, binding specifically to gp41 and gp120 due to the amino acids basic sequence, it was shown that SB105-A10 could inhibit HIV-1 infection through binding with molecular targets on the cell membrane such as HSPG.51 SB105-A10 binds to HS on the cell membrane although removal of HS did not result in a complete inhibition of the binding of SB105-A10 to the cell membrane, suggesting that the dendrimer may also display a second mechanism of action not associated with HS. Through SPR assays it was demonstrated a strong binding affinity between gp41 and peptide derivatized-dendrimers. The binding pocket of g41 associated with this interaction is strongly involved in the fusion between the virus and the cell surface.52
Several glycosphingolipids, such as galactosyl ceramide (GalCer) and its 3′-sulfated derivative, sulfatide (SGalCer), have been reported to be the receptors that allow HIV-1 infection in CD4 deficient neural, colonic and vaginal cells.53–55 SGalCer was determined as the best ligand or cofactor for HIV-1 gp120, among different GSLs, for each of the rgp120s and HIV-1 tested, regardless of cellular tropism.56 It was postulated that charge alone would be the responsible for this higher affinity. However, it is expected that the negatively charged, sialylated GSLs bind rgp120 with at lest equal avidity. Therefore, it can be concluded that SGalCer sulfate groups might be carry out some interaction which lead SGalCer to be the favorite GSL ligand for gp120.56 We can hypostatize that the ability of those SGalCer to bind to gp120 due to their sulfated groups could be associated with HS, these sulfated groups could be acting as HS analogs increasing the affinity of gp120 for SGalCer compared to other GSLs. Consolidating this hypothesis the fact the inhibitory effects of these polysulfated polysaccharides comes from their interaction with the positively charged V3 loops on HIV-1 gp120,57 which, as mentioned earlier, is highly associated with HS.39,40
Based on these premises, polysulfated galactose functionalized glycodendrimer (PS Gal 64mer) that contained two sulfate groups per galactose was synthesized and evaluated against HIV-1 infection. The ability of the PS Gal 64mer to inhibit HIV-1 infection was compared to that of dextran sulfate (DxS), a known potent inhibitor of X4 HIV-1 infectivity. The results show that the PS Gal 64mer inhibited HIV-1 isolates as well as DxS with EC50 values in the nanomolar range.56
Other dendrimers have presented dual acting mechanism such as four generation-PAMAM 24 branched-naphthalene disulfonic surface groups ended SPL2923 and the 32 phenyldicarboxylic acid SLP6195. They were evaluated against in different strains HIV-1. It was shown that these dendrimers were able to perform their antiviral activity against HIV-1 infection by blocking virus–cell binding via gp120. Despite their primary mechanism of action dendrimers must be allocated to interference with the gp120-driven virus–cell-binding process. The ability of these dendrimers to penetrate into the cell and block two intracellular stages of the replication cycle of HIV-1: reverse transcription and integration was demonstrated.58
Due to the good results obtained with the previous dendrimers, L-lysine branched dendrimers, with higher stability and inhibitory potency compared to these synthetized using polyamidoamine (PAMAM) linkages approaches, were evaluated against HIV-1 infection as they were against HSV-2.59 The inhibitory activities in vitro of several dendrimers with 1 to 5 L-lysine branches and fully capped with one of seven different surface groups were evaluated. Second (SPL7115) and fourth (SPL7013) generation dendrimers with naphthalene disulfonic acid (DNAA) surface groups were selected due to their potent broad-spectrum antiviral activity and potential to block cell-to-cell transmission of HIV-1.25 Mechanism of action studies and modeling studies showed that SPL7013 blocks the entry of both X4 and R5 HIV-1 strains with equal potency and inhibits X4 and R5 HIV-1 envelope mediated cell-to-cell fusion through electrostatic interactions of dendrimers with gp120.
Molecular modeling also suggested that electrostatic interaction of both dendrimers with the highly positively charged V3 loop on gp120 of X4 HIV-1 strains and interactions with basic residues in gp120 of both X4 and R5 strains.25 It has been suggested that the difference between viral isolates could be explained by the ability of sulfated compounds to bind with higher avidity to the V3 loop of X4 HIV strains than that shown when they bind to V3 loops of R5 HIV-1, which is highly associated with HS–gp120 interaction.44
In addition, the same study reported that dendrimer anionic surface charge as well as size is important for HIV-1 and HSV inhibition. Larger dendrimers could cause steric hindrance between viral gp120 and host cell receptor interactions. However, later on it was shown that SPL7013 mechanism does not involve disruption of the viral particle or loss of gp120 from the viral surface. SPL7013 might be binding tightly to HIV-1 envelope proteins, physically blocking attachment of the virions to the cell receptors, or preventing interactions with viral envelope proteins that lead to envelope conformational changes that abrogate viral infectivity.60
Over the years, several polyanionic carbosilane dendrimers based on a silica atom core have been reported as potent HIV infection inhibitors.61–66 Sulfated and naphthylsulfonated-decorated dendrimers: first-generation G1-S4, third-generation G3-S16 and second-generation G2-NF16, as well as, carbosilane dendrimers bearing sulfonate or carboxylate groups at their periphery synthesized by thiol–ene or Michael addition chemistry, second-generation sulfonate-functionalized G2-S16 and second-generation via thiol–ene-synthesized G2-STE16 were evaluated against HIV-1 infection.62,64,66
These nanosystems present a C and Si based skeleton and they differ from other dendrimers in the high apolarity of their central core and the high mobility of their peripheral branches.
Carbosilane dendrimers' mode of action is a multifactorial process targeting several proteins from viral envelope as well as from host cell. G3-S16 and G2-NF16 are unspecific polyanionic compounds and electrostatic interactions between their periphery functional groups and proteins of the viral envelope, such as gp120, play a major role inhibiting viral infection at fusion step, blocking gp120/CD4 interaction. In addition, G3-S16 and G2-NF16 inhibit cell-to-cell HIV transmission and difficult infectious synapse formation.66
Evaluation of the mechanism of action of G2-S16 showed that this sulfonated dendrimer blocked gp120/CD4 mediated membrane fusion. This inhibition did not involve degradation integrity of the viral membrane by destabilizing the core-membrane linkage or by shedding of gp120. Several computer models of G2-S16 confirmed that G2-S16 forms more stable, and irreversible, complexes with gp120 than CD4, therefore, abrogate viral infectivity.61,62,64
In addition to the capacity to inhibit HIV-1 sexual transmission, G2-S16, G2-STE16 and G3-S16 showed anti-HIV-2 activity at early stages of viral infection inactivating the virus and blocking the binding of gp120 to CD4, and the HIV-2 entry.61
Although the relationship between HS and carbosilane dendrimers is not currently proven, the last results obtained with these compounds and HSV-2 are reminiscent of greater relevance of this HSPGs in the inhibition of HIV-1. For example, sulfated groups of G1-S4 and G3-S16 could be acting as analogs of HS and, therefore, reducing HIV-1 infection.
Summing up, dendrimers able to stymie HS interaction with HIV-1 will result in a decreased transmission of HIV-1 to dendritic cells, macrophages and T cells, due to the inhibition of HS ability of spermatozoa to agglutinate HIV-1 particles and its capacity to sequester viral particles, therefore avoiding viral translocation across epithelial barriers.67,68 This ability of HS to enhance viral infection via agglutination is similar to that described of the seminal amyloids of the semen to bind to HIV virions and efficiently enhance and accelerate their attachment to target cells,69–72 which, recently, has been described as a critical point in the development of new topical microbicides and their ability to surpass clinical trial.73
Role of heparan sulfate in dendrimers against HCV infection
HCV infection is a major cause of chronic hepatitis worldwide, leading to steatosis, liver cirrhosis and hepatocellular carcinoma.74 HCV entry into the host cell is a multistep process. The main hepatocyte receptors involved in cell entry are tetraspanin CD81,75 human SR-BI76 and tight junction molecules claudin-1 (ref. 77) and occluding.78 The cell surface glycosaminoglycans, such as HS, play a major role mediating the initial virus attachment.79–81Viral proteins C, E1, E2 and p7, among others, are necessary for viral infection.82–84 Although it was initially thought that they joined them directly to cellular glycosaminoglycans, recently, it has been shown that this union is mediated by apolipoprotein E (ApoE). ApoE mediates attachment of the surface of the HCV envelope through a specific interaction with cell surface HS but not via other known HCV receptors and/or coreceptors.85,86 This interaction with highly sulfated HS on target cells is not simply the result of charge interactions but requires a specific HS structure. This interaction with target cells is not simply due to the charge interactions but requires a specific structure of the HS, in particular, specific HS involving N- and 6-O-sulfate groups.85
Recently, a screening of several polyanionic carbosilane dendrimers was performed to indentify non-toxic antiviral compounds targeting different steps of the HCV lifecycle. These dendrimers had already demonstrated potent and broad-spectrum anti-HIV-1 and/or HSV-2 activity in vitro and in vivo. Four sulfonated polyanionic carbosilane dendrimers (G2-S24P, G2-STE16, G2-S16 and G3-STE24P) and two carboxylate coated dendrimers (G2-CTE16 and G3-CTE24P) were selected due to their inhibitory values, genotype spectrum and step targeted inhibition. It was demonstrated that polyanionic carbosilane dendrimers inhibit infection by genotype 2a HCVtcp and HCVpp of major genotypes (1, 2, 3 and 4).87 It was shown that sulfonate coated dendrimers bind to the cationic residues of the virus which bind to HS and act as molecular decoys that prevent recognition of HS in the host cell. There were two theories on the ability of the carboxylate polyanionic carbosilane dendrimers postulated, the first one affirms that these dendrimers could be acting similarly to the sulfonates dendrimers, whereas the second theory postulates that a conformational change occurs in the virus avoiding the recognition of the cell HS and consequently the viral entry.
IHS has been shown to play an important role in Hepatitis B Virus (HBV) infection. HBV infection requires the initial attachment to the carbohydrate side chains of hepatocyte-associated HS (HSPGs) as attachment receptors.88 Specifically, HS D-glucosaminyl 3-O-sulfotransferase 3 B1 plays a major role in the initial attachment. However, it has not yet be reported that dendrimers are able to inhibit HBV infection, the fact that a common mechanism of entry, such as HS, with other diseases, suggests that perhaps the use of dendrimers able to inhibit successfully viral infection at HS level could be a good candidates against HBV.88
Role of heparan sulphate in dendrimers against human papillomavirus infection
HPV is the STI with the highest prevalence in the world. HPVs are classified as “low-risk” or “high-risk” types according to their association with cervical cancer. Infection with high-risk HPV types (primarily types 16, 18, 31, and 45) can cause cervical cell abnormalities that are precursors to cancer.89Currently, the FDA has approved three vaccines to prevent infection with HPV: Gardasil, Gardasil 9 (Merck and Co., Inc., Whitehouse Station, NJ)90,91 and Cervarix (GlaxoSmith-Kline Biologicals, Rixensart, Belgium).92 The three vaccines prevent infection by HPV type 16 and 18, two of the HPV high risk of causing about 70% of invasive cervical cancers and even higher percentage of some other related cancers HPV.93,94 Due to low accessibility and high price of these vaccines in developing countries, the development of alternative therapies such as microbicides would be a good option.
The interaction between the virion proteins and cell surface HSPGs mediates the initial attachment of HPV to the target cell.95,96 This interaction appears to be mediated by basic domains of viral proteins L1 and L2 (6, 35) and the negatively charged sulfated/carboxyl groups of the HS chains (Fig. 4). This interaction represents a major target for the development of a microbicide able to disrupt the sexual transmission of HPV among other viruses, such as HSV-2 or HIV-1.
 | ||
| Fig. 4 HCV entry. HCV LVPs attach to the cell surface by interaction with HSPG, LDLR and SR-BI. (A) HCV binds to HSPGs on the membrane exposed after disruption. (B) This induces a conformational change exposing a site on L2 susceptible to proprotein convertase cleavage. (C) After L2 cleavage, an L2 neutralizing epitope (CD81 binding site) is exposed and a previously unexposed region of L1 binds to a secondary receptor on the epithelial cells. This interaction leads to activation of transduction which promotes lateral movement and clathrin-mediated endocytosis. | ||
Right now, there is only a dendrimer, SB105, whose effectiveness against HPV has been assessed. As noted earlier, this dendrimer also disrupt the sexual transmission of HIV and HSV-2.17,51 SB105-A10 was found to inhibit HPV attachment to the cellular HSPGs. Although, further research is needed the binding of SB105-A10 to HSPGs is required for its antiviral activity. SB105-A10 inhibits HPV infection when it was added to the cells 2 hours before the infection and the washed. SB105-A10 did not prevent infection by the human rotavirus Wa, a virus that does not need HSPGs for attachment, showing that its anti-HPV-16 potential is dependent on its interaction with the cell surface.97
SB105-A10 has shown its ability to halt most STIs associated with HS. However, it is also noteworthy its ability to inhibit other viral infection associated with HS, such as human cytomegalovirus (HCMV)19 and human respiratory syncytial virus (RSV).98
Conflict of interest
No conflicts of interest were found.Acknowledgements
Financial support: this work has been (partially) funded by the RD12/0017/0037, project as part of the project as part of the Acción Estratégica en Salud, Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica 2008–2011 and cofinanced by Instituto de Salud Carlos III (Subdirección General de Evaluación) and Fondo Europeo de Desarrollo Regional (FEDER), RETIC PT13/0010/0028, Fondo de Investigacion Sanitaria (FIS) (grant number PI13/02016), Comunidad de Madrid (S-2010/BMD-2332), CYTED 214RT0482. CIBER-BBN is an initiative funded by the VI National R&D&i Plan 2008–2011, IniciativaIngenio 2010, the Consolider Program, and CIBER Actions and financed by the Instituto de Salud Carlos III with assistance from the European Regional Development Fund. Marek Maly gratefully acknowledges project GA15-05903S. This work was supported partially by a Marie Curie International Research Staff Exchange Scheme Fellowship within the 7th European Community Framework Program, project No. PIRSES-GA-2012-316730 NANOGENE, co-financed by the Polish Ministry of Science and Higher Education (grant No. W21/7PR/2013).References
- K. Owusu-Edusei Jr, H. W. Chesson, T. L. Gift, G. Tao, R. Mahajan, M. C. Ocfemia and C. K. Kent, Sex. Transm. Dis., 2013, 40, 197–201 CrossRef PubMed.
- C. L. Satterwhite, E. Torrone, E. Meites, E. F. Dunne, R. Mahajan, M. C. Ocfemia, J. Su, F. Xu and H. Weinstock, Sex. Transm. Dis., 2013, 40, 187–193 CrossRef PubMed.
- A. Rompalo, J. Clin. Invest., 2011, 121, 4580–4583 CrossRef CAS PubMed.
- E. E. Freeman, H. A. Weiss, J. R. Glynn, P. L. Cross, J. A. Whitworth and R. J. Hayes, AIDS, 2006, 20, 73–83 CrossRef PubMed.
- J. R. Bishop, M. Schuksz and J. D. Esko, Nature, 2007, 446, 1030–1037 CrossRef CAS PubMed.
- J. D. Esko and U. Lindahl, J. Clin. Invest., 2001, 108, 169–173 CrossRef CAS PubMed.
- Y. Huang, Y. Mao, C. Zong, C. Lin, G. J. Boons and J. Zaia, Anal. Chem., 2015, 87, 592–600 CrossRef CAS PubMed.
- K. J. Looker, A. S. Magaret, K. M. Turner, P. Vickerman, S. L. Gottlieb and L. M. Newman, PLoS One, 2015, 10, e114989 Search PubMed.
- R. Gupta, T. Warren and A. Wald, Lancet, 2007, 370, 2127–2137 CrossRef.
- C. Johnston, M. Saracino, S. Kuntz, A. Magaret, S. Selke, M. L. Huang, J. T. Schiffer, D. M. Koelle, L. Corey and A. Wald, Lancet, 2012, 379, 641–647 CrossRef CAS.
- C. Johnston, D. M. Koelle and A. Wald, Vaccine, 2014, 32, 1553–1560 CrossRef CAS PubMed.
- B. E. Thacker, D. Xu, R. Lawrence and J. D. Esko, Matrix Biol., 2014, 35, 60–72 CrossRef CAS PubMed.
- J. Akhtar and D. Shukla, FEBS J., 2009, 276, 7228–7236 CrossRef CAS PubMed.
- P. G. Spear, Cell. Microbiol., 2004, 6, 401–410 CrossRef CAS PubMed.
- M. J. Lehmann, N. M. Sherer, C. B. Marks, M. Pypaert and W. Mothes, J. Cell Biol., 2005, 170, 317–325 CrossRef CAS PubMed.
- M. Schelhaas, H. Ewers, M. L. Rajamaki, P. M. Day, J. T. Schiller and A. Helenius, PLoS Pathog., 2008, 4, e1000148 Search PubMed.
- A. Luganini, S. F. Nicoletto, L. Pizzuto, G. Pirri, A. Giuliani, S. Landolfo and G. Gribaudo, Antimicrob. Agents Chemother., 2011, 55, 3231–3239 CrossRef CAS PubMed.
- A. Pini, A. Giuliani, C. Falciani, Y. Runci, C. Ricci, B. Lelli, M. Malossi, P. Neri, G. M. Rossolini and L. Bracci, Antimicrob. Agents Chemother., 2005, 49, 2665–2672 CrossRef CAS PubMed.
- A. Luganini, A. Giuliani, G. Pirri, L. Pizzuto, S. Landolfo and G. Gribaudo, Antiviral Res., 2010, 85, 532–540 CrossRef CAS PubMed.
- N. Bourne, L. R. Stanberry, E. R. Kern, G. Holan, B. Matthews and D. I. Bernstein, Antimicrob. Agents Chemother., 2000, 44, 2471–2474 CrossRef CAS PubMed.
- Y. Gong, B. Matthews, D. Cheung, T. Tam, I. Gadawski, D. Leung, G. Holan, J. Raff and S. Sacks, Antiviral Res., 2002, 55, 319–329 CrossRef CAS PubMed.
- D. I. Bernstein, L. R. Stanberry, S. Sacks, N. K. Ayisi, Y. H. Gong, J. Ireland, R. J. Mumper, G. Holan, B. Matthews, T. McCarthy and N. Bourne, Antimicrob. Agents Chemother., 2003, 47, 3784–3788 CrossRef CAS PubMed.
- R. Rupp, S. L. Rosenthal and L. R. Stanberry, Int. J. Nanomed., 2007, 2, 561–566 CAS.
- E. Gong, B. Matthews, T. McCarthy, J. Chu, G. Holan, J. Raff and S. Sacks, Antiviral Res., 2005, 68, 139–146 CrossRef CAS PubMed.
- D. Tyssen, S. A. Henderson, A. Johnson, J. Sterjovski, K. Moore, J. La, M. Zanin, S. Sonza, P. Karellas, M. P. Giannis, G. Krippner, S. Wesselingh, T. McCarthy, P. R. Gorry, P. A. Ramsland, R. Cone, J. R. Paull, G. R. Lewis and G. Tachedjian, PLoS One, 2010, 5, e12309 Search PubMed.
- S. Galdiero, A. Falanga, M. Vitiello, M. D'Isanto, M. Cantisani, A. Kampanaraki, E. Benedetti, H. Browne and M. Galdiero, Peptides, 2008, 29, 1461–1471 CrossRef CAS PubMed.
- S. Galdiero, A. Falanga, M. Vitiello, L. Raiola, R. Fattorusso, H. Browne, C. Pedone, C. Isernia and M. Galdiero, J. Biol. Chem., 2008, 283, 29993–30009 CrossRef CAS PubMed.
- S. Galdiero, A. Falanga, M. Vitiello, L. Raiola, L. Russo, C. Pedone, C. Isernia and M. Galdiero, J. Biol. Chem., 2010, 285, 17123–17136 CrossRef CAS PubMed.
- R. Tarallo, T. P. Carberry, A. Falanga, M. Vitiello, S. Galdiero, M. Galdiero and M. Weck, Int. J. Nanomed., 2013, 8, 521–534 CrossRef PubMed.
- R. Ceña-Diez, E. Vacas-Córdoba, P. García-Broncano, F. de la Mata, R. Gómez, M. Maly and M. Muñoz-Fernández, Int. J. Nanomed., 2016 Search PubMed.
- F. C. Bender, J. C. Whitbeck, H. Lou, G. H. Cohen and R. J. Eisenberg, J. Virol., 2005, 79, 11588–11597 CrossRef CAS PubMed.
- S. H. Basha, D. Talluri and N. P. Raminni, BMC Complementary Altern. Med., 2013, 13, 85 CrossRef PubMed.
- A. Jacquet, M. Haumont, D. Chellun, M. Massaer, F. Tufaro, A. Bollen and P. Jacobs, Virus Res., 1998, 53, 197–207 CrossRef CAS PubMed.
- D. Shukla and P. G. Spear, J. Clin. Invest., 2001, 108, 503–510 CrossRef CAS PubMed.
- T. Compton, D. M. Nowlin and N. R. Cooper, Virology, 1993, 193, 834–841 CrossRef CAS PubMed.
- J. Neyts, R. Snoeck, D. Schols, J. Balzarini, J. D. Esko, A. Van Schepdael and E. De Clercq, Virology, 1992, 189, 48–58 CrossRef CAS PubMed.
- E. De Clercq, Curr. Opin. Pharmacol., 2010, 10, 507–515 CrossRef CAS PubMed.
- M. M. Lederman, R. Jump, H. A. Pilch-Cooper, M. Root and S. F. Sieg, Retrovirology, 2008, 5, 116 CrossRef PubMed.
- R. Wyatt, J. Moore, M. Accola, E. Desjardin, J. Robinson and J. Sodroski, J. Virol., 1995, 69, 5723–5733 CAS.
- P. D. Kwong, R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski and W. A. Hendrickson, Nature, 1998, 393, 648–659 CrossRef CAS PubMed.
- S. Basmaciogullari, G. J. Babcock, D. Van Ryk, W. Wojtowicz and J. Sodroski, J. Virol., 2002, 76, 10791–10800 CrossRef CAS PubMed.
- J. C. Tilton and R. W. Doms, Antiviral Res., 2010, 85, 91–100 CrossRef CAS PubMed.
- I. Mondor, S. Ugolini and Q. J. Sattentau, J. Virol., 1998, 72, 3623–3634 CAS.
- M. Moulard, H. Lortat-Jacob, I. Mondor, G. Roca, R. Wyatt, J. Sodroski, L. Zhao, W. Olson, P. D. Kwong and Q. J. Sattentau, J. Virol., 2000, 74, 1948–1960 CrossRef CAS PubMed.
- L. de Witte, Y. Zoughlami, B. Aengeneyndt, G. David, Y. van Kooyk, L. Gissmann and T. B. Geijtenbeek, Immunobiology, 2007, 212, 679–691 CrossRef CAS PubMed.
- P. Gallay, Microbes Infect., 2004, 6, 617–622 CrossRef CAS PubMed.
- Z. Wu, Z. Chen and D. M. Phillips, J. Infect. Dis., 2003, 188, 1473–1482 CrossRef PubMed.
- E. G. Argyris, E. Acheampong, G. Nunnari, M. Mukhtar, K. J. Williams and R. J. Pomerantz, J. Virol., 2003, 77, 12140–12151 CrossRef CAS PubMed.
- M. Patel, M. Yanagishita, G. Roderiquez, D. C. Bou-Habib, T. Oravecz, V. C. Hascall and M. A. Norcross, AIDS Res. Hum. Retroviruses, 1993, 9, 167–174 CrossRef CAS PubMed.
- A. C. Saphire, M. D. Bobardt, Z. Zhang, G. David and P. A. Gallay, J. Virol., 2001, 75, 9187–9200 CrossRef CAS PubMed.
- I. Bon, D. Lembo, M. Rusnati, A. Clo, S. Morini, A. Miserocchi, A. Bugatti, S. Grigolon, G. Musumeci, S. Landolfo, M. C. Re and D. Gibellini, PLoS One, 2013, 8, e76482 CAS.
- D. K. Chang and C. S. Hsu, Antiviral Res., 2007, 74, 51–58 CrossRef CAS PubMed.
- Y. Furuta, K. Eriksson, B. Svennerholm, P. Fredman, P. Horal, S. Jeansson, A. Vahlne, J. Holmgren and C. Czerkinsky, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12559–12563 CrossRef CAS.
- J. M. Harouse, R. G. Collman and F. Gonzalez-Scarano, J. Virol., 1995, 69, 7383–7390 CAS.
- N. Yahi, S. Baghdiguian, H. Moreau and J. Fantini, J. Virol., 1992, 66, 4848–4854 CAS.
- R. D. Kensinger, B. J. Catalone, F. C. Krebs, B. Wigdahl and C. L. Schengrund, Antimicrob. Agents Chemother., 2004, 48, 1614–1623 CrossRef CAS PubMed.
- P. P. Jagodzinski, J. Wustner, D. Kmieciak, T. J. Wasik, A. Fertala, A. L. Sieron, M. Takahashi, T. Tsuji, T. Mimura, M. S. Fung, M. K. Gorny, M. Kloczewiak, Y. Kaneko and D. Kozbor, Virology, 1996, 226, 217–227 CrossRef CAS PubMed.
- M. Witvrouw, V. Fikkert, W. Pluymers, B. Matthews, K. Mardel, D. Schols, J. Raff, Z. Debyser, E. De Clercq, G. Holan and C. Pannecouque, Mol. Pharmacol., 2000, 58, 1100–1108 CAS.
- T. D. McCarthy, P. Karellas, S. A. Henderson, M. Giannis, D. F. O'Keefe, G. Heery, J. R. Paull, B. R. Matthews and G. Holan, Mol. Pharm., 2005, 2, 312–318 CrossRef CAS PubMed.
- S. Telwatte, K. Moore, A. Johnson, D. Tyssen, J. Sterjovski, M. Aldunate, P. R. Gorry, P. A. Ramsland, G. R. Lewis, J. R. Paull, S. Sonza and G. Tachedjian, Antiviral Res., 2011, 90, 195–199 CrossRef CAS PubMed.
- V. Briz, D. Sepulveda-Crespo, A. R. Diniz, P. Borrego, B. Rodes, F. J. de la Mata, R. Gomez, N. Taveira and M. A. Munoz-Fernandez, Nanoscale, 2015, 7, 14669–14683 RSC.
- L. Chonco, M. Pion, E. Vacas, B. Rasines, M. Maly, M. J. Serramia, L. Lopez-Fernandez, J. De la Mata, S. Alvarez, R. Gomez and M. A. Munoz-Fernandez, J. Controlled Release, 2012, 161, 949–958 CrossRef CAS PubMed.
- J. Sanchez-Rodriguez, L. Diaz, M. Galan, M. Maly, R. Gomez, F. Javier de la Mata, J. L. Jimenez and M. A. Munoz-Fernandez, J. Biomed. Nanotechnol., 2015, 11, 1783–1798 CrossRef CAS PubMed.
- D. Sepulveda-Crespo, R. Gomez, F. J. De La Mata, J. L. Jimenez and M. A. Munoz-Fernandez, Nanomedicine, 2015, 11, 1481–1498 CAS.
- D. Sepulveda-Crespo, M. J. Serramia, A. M. Tager, V. Vrbanac, R. Gomez, F. J. De La Mata, J. L. Jimenez and M. A. Munoz-Fernandez, Nanomedicine, 2015, 11, 1299–1308 CAS.
- E. Vacas Cordoba, E. Arnaiz, M. Relloso, C. Sanchez-Torres, F. Garcia, L. Perez-Alvarez, R. Gomez, F. J. de la Mata, M. Pion and M. A. Munoz-Fernandez, AIDS, 2013, 27, 1219–1229 CrossRef CAS PubMed.
- A. Ceballos, F. Remes Lenicov, J. Sabatte, C. Rodriguez Rodrigues, M. Cabrini, C. Jancic, S. Raiden, M. Donaldson, R. Agustin Pasqualini Jr, C. Marin-Briggiler, M. Vazquez-Levin, F. Capani, S. Amigorena and J. Geffner, J. Exp. Med., 2009, 206, 2717–2733 CrossRef CAS PubMed.
- E. Crublet, J. P. Andrieu, R. R. Vives and H. Lortat-Jacob, J. Biol. Chem., 2008, 283, 15193–15200 CrossRef CAS PubMed.
- J. Munch, E. Rucker, L. Standker, K. Adermann, C. Goffinet, M. Schindler, S. Wildum, R. Chinnadurai, D. Rajan, A. Specht, G. Gimenez-Gallego, P. C. Sanchez, D. M. Fowler, A. Koulov, J. W. Kelly, W. Mothes, J. C. Grivel, L. Margolis, O. T. Keppler, W. G. Forssmann and F. Kirchhoff, Cell, 2007, 131, 1059–1071 CrossRef PubMed.
- N. R. Roan, J. A. Muller, H. Liu, S. Chu, F. Arnold, C. M. Sturzel, P. Walther, M. Dong, H. E. Witkowska, F. Kirchhoff, J. Munch and W. C. Greene, Cell Host Microbe, 2011, 10, 541–550 CAS.
- F. Arnold, J. Schnell, O. Zirafi, C. Sturzel, C. Meier, T. Weil, L. Standker, W. G. Forssmann, N. R. Roan, W. C. Greene, F. Kirchhoff and J. Munch, J. Virol., 2012, 86, 1244–1249 CrossRef CAS PubMed.
- S. M. Usmani, O. Zirafi, J. A. Muller, N. L. Sandi-Monroy, J. K. Yadav, C. Meier, T. Weil, N. R. Roan, W. C. Greene, P. Walther, K. P. Nilsson, P. Hammarstrom, R. Wetzel, C. D. Pilcher, F. Gagsteiger, M. Fandrich, F. Kirchhoff and J. Munch, Nat. Commun., 2014, 5, 3508 Search PubMed.
- O. Zirafi, K. A. Kim, N. R. Roan, S. F. Kluge, J. A. Muller, S. Jiang, B. Mayer, W. C. Greene, F. Kirchhoff and J. Munch, Sci. Transl. Med., 2014, 6, 262ra157 CrossRef PubMed.
- C. W. Shepard, L. Finelli and M. J. Alter, Lancet Infect. Dis., 2005, 5, 558–567 CrossRef PubMed.
- P. Pileri, Y. Uematsu, S. Campagnoli, G. Galli, F. Falugi, R. Petracca, A. J. Weiner, M. Houghton, D. Rosa, G. Grandi and S. Abrignani, Science, 1998, 282, 938–941 CrossRef CAS PubMed.
- E. Scarselli, H. Ansuini, R. Cerino, R. M. Roccasecca, S. Acali, G. Filocamo, C. Traboni, A. Nicosia, R. Cortese and A. Vitelli, EMBO J., 2002, 21, 5017–5025 CrossRef CAS PubMed.
- M. J. Evans, T. von Hahn, D. M. Tscherne, A. J. Syder, M. Panis, B. Wolk, T. Hatziioannou, J. A. McKeating, P. D. Bieniasz and C. M. Rice, Nature, 2007, 446, 801–805 CrossRef CAS PubMed.
- S. Liu, W. Yang, L. Shen, J. R. Turner, C. B. Coyne and T. Wang, J. Virol., 2009, 83, 2011–2014 CrossRef CAS PubMed.
- H. Barth, E. K. Schnober, F. Zhang, R. J. Linhardt, E. Depla, B. Boson, F. L. Cosset, A. H. Patel, H. E. Blum and T. F. Baumert, J. Virol., 2006, 80, 10579–10590 CrossRef CAS PubMed.
- K. Morikawa, Z. Zhao, T. Date, M. Miyamoto, A. Murayama, D. Akazawa, J. Tanabe, S. Sone and T. Wakita, J. Med. Virol., 2007, 79, 714–723 CrossRef CAS PubMed.
- H. Barth, C. Schafer, M. I. Adah, F. Zhang, R. J. Linhardt, H. Toyoda, A. Kinoshita-Toyoda, T. Toida, T. H. Van Kuppevelt, E. Depla, F. Von Weizsacker, H. E. Blum and T. F. Baumert, J. Biol. Chem., 2003, 278, 41003–41012 CrossRef CAS PubMed.
- V. Jirasko, R. Montserret, N. Appel, A. Janvier, L. Eustachi, C. Brohm, E. Steinmann, T. Pietschmann, F. Penin and R. Bartenschlager, J. Biol. Chem., 2008, 283, 28546–28562 CrossRef CAS PubMed.
- C. T. Jones, C. L. Murray, D. K. Eastman, J. Tassello and C. M. Rice, J. Virol., 2007, 81, 8374–8383 CrossRef CAS PubMed.
- E. Steinmann, F. Penin, S. Kallis, A. H. Patel, R. Bartenschlager and T. Pietschmann, PLoS Pathog., 2007, 3, e103 Search PubMed.
- Y. Xu, P. Martinez, K. Seron, G. Luo, F. Allain, J. Dubuisson and S. Belouzard, J. Virol., 2015, 89, 3846–3858 CrossRef CAS PubMed.
- J. Jiang, W. Cun, X. Wu, Q. Shi, H. Tang and G. Luo, J. Virol., 2012, 86, 7256–7267 CrossRef CAS PubMed.
- D. Sepúlveda-Crespo, J. Jiménez, R. Gómez, F. De La Mata, P. Majano, M. Muñoz-Fernández and P. Gastaminza, Nanomedicine, 2016 Search PubMed.
- A. Schulze, P. Gripon and S. Urban, Hepatology, 2007, 46, 1759–1768 CrossRef CAS PubMed.
- F. X. Bosch and S. de Sanjose, J. Natl. Cancer Inst. Monogr., 2003, 3–13 CrossRef.
- L. L. Villa, R. L. Costa, C. A. Petta, R. P. Andrade, K. A. Ault, A. R. Giuliano, C. M. Wheeler, L. A. Koutsky, C. Malm, M. Lehtinen, F. E. Skjeldestad, S. E. Olsson, M. Steinwall, D. R. Brown, R. J. Kurman, B. M. Ronnett, M. H. Stoler, A. Ferenczy, D. M. Harper, G. M. Tamms, J. Yu, L. Lupinacci, R. Railkar, F. J. Taddeo, K. U. Jansen, M. T. Esser, H. L. Sings, A. J. Saah and E. Barr, Lancet Oncol., 2005, 6, 271–278 CrossRef PubMed.
- C. Printz, Cancer, 2015, 121, 1156–1157 CrossRef PubMed.
- D. M. Harper, E. L. Franco, C. Wheeler, D. G. Ferris, D. Jenkins, A. Schuind, T. Zahaf, B. Innis, P. Naud, N. S. De Carvalho, C. M. Roteli-Martins, J. Teixeira, M. M. Blatter, A. P. Korn, W. Quint and G. Dubin, Lancet, 2004, 364, 1757–1765 CrossRef CAS.
- A. K. Chaturvedi, E. A. Engels, R. M. Pfeiffer, B. Y. Hernandez, W. Xiao, E. Kim, B. Jiang, M. T. Goodman, M. Sibug-Saber, W. Cozen, L. Liu, C. F. Lynch, N. Wentzensen, R. C. Jordan, S. Altekruse, W. F. Anderson, P. S. Rosenberg and M. L. Gillison, J. Clin. Oncol., 2011, 29, 4294–4301 CrossRef PubMed.
- M. L. Gillison, A. K. Chaturvedi and D. R. Lowy, Cancer, 2008, 113, 3036–3046 CrossRef PubMed.
- S. Shafti-Keramat, A. Handisurya, E. Kriehuber, G. Meneguzzi, K. Slupetzky and R. Kirnbauer, J. Virol., 2003, 77, 13125–13135 CrossRef CAS PubMed.
- T. Giroglou, L. Florin, F. Schafer, R. E. Streeck and M. Sapp, J. Virol., 2001, 75, 1565–1570 CrossRef CAS PubMed.
- M. Donalisio, M. Rusnati, A. Civra, A. Bugatti, D. Allemand, G. Pirri, A. Giuliani, S. Landolfo and D. Lembo, Antimicrob. Agents Chemother., 2010, 54, 4290–4299 CrossRef CAS PubMed.
- M. Donalisio, M. Rusnati, V. Cagno, A. Civra, A. Bugatti, A. Giuliani, G. Pirri, M. Volante, M. Papotti, S. Landolfo and D. Lembo, Antimicrob. Agents Chemother., 2012, 56, 5278–5288 CrossRef CAS PubMed.
- C. D. O'Donnell, M. Kovacs, J. Akhtar, T. Valyi-Nagy and D. Shukla, Virology, 2010, 397, 389–398 CrossRef PubMed.
- D. Shukla, J. Liu, P. Blaiklock, N. W. Shworak, X. Bai, J. D. Esko, G. H. Cohen, R. J. Eisenberg, R. D. Rosenberg and P. G. Spear, Cell, 1999, 99, 13–22 CrossRef CAS PubMed.
- N. Cheshenko and B. C. Herold, J. Gen. Virol., 2002, 83, 2247–2255 CrossRef CAS PubMed.
- E. M. Borst, L. Standker, K. Wagner, T. F. Schulz, W. G. Forssmann and M. Messerle, Antimicrob. Agents Chemother., 2013, 57, 4751–4760 CrossRef CAS PubMed.
- K. M. Johnson, R. C. Kines, J. N. Roberts, D. R. Lowy, J. T. Schiller and P. M. Day, J. Virol., 2009, 83, 2067–2074 CrossRef CAS PubMed.
- P. M. Matos, D. Andreu, N. C. Santos and R. Gutierrez-Gallego, Arch. Virol., 2014, 159, 555–560 CrossRef CAS PubMed.
- S. Fadel and A. Eley, J. Med. Microbiol., 2008, 57, 1058–1061 CrossRef CAS PubMed.
- J. F. Alderete and J. B. Baseman, Genitourin. Med., 1989, 65, 177–182 CAS.
- E. Freissler, A. Meyer auf der Heyde, G. David, T. F. Meyer and C. Dehio, Cell. Microbiol., 2000, 2, 69–82 CrossRef CAS PubMed.
- V. Tiwari, E. Maus, I. M. Sigar, K. H. Ramsey and D. Shukla, Glycobiology, 2012, 22, 1402–1412 CrossRef CAS PubMed.
- M. P. Hoffman and C. G. Haidaris, Infect. Immun., 1994, 62, 828–836 CAS.
| This journal is © The Royal Society of Chemistry 2016 |