
Graphene based magnetic nanocomposites as versatile carriers for high yield immobilization and stabilization of β-galactosidase†
Maryam Khana,
Qayyum Husain*a and
Alim Husain Naqvib
aDepartment of Biochemistry, Faculty of Life Sciences, Aligarh Muslim University, Aligarh, 202002, UP, India. E-mail: qayyumbiochem@gmail.com; Tel: +91-9897701792
bCentre of Excellence in Materials Science (Nanomaterials), Department of Applied Physics, Z. H. College of Engineering & Technology, Aligarh Muslim University, Aligarh, 202002, UP, India
First published on 27th May 2016
Abstract
The present study demonstrates an efficient method for high yield immobilization of Aspergillus oryzae β-galactosidase onto graphene-iron oxide nanocomposites (Gr@Fe3O4 NCs) by a simple adsorption mechanism. The synthesized nanocomposites were characterized by X-ray diffraction and FT-Raman spectroscopy, and by vibrating sample magnetometry. The binding of the enzyme onto the nanocomposites was confirmed by transmission electron microscopy, scanning electron microscopy, and Fourier transform-infrared spectroscopy. The bound enzyme showed 90% immobilization yield. The adsorbed and free enzyme both exhibited the same pH-optima at pH 4.5. However, the immobilized enzyme showed enhanced pH stability toward acidic and basic extremes, as well as increased temperature resistance as compared to native β-galactosidase. The Michaelis constant, Km decreased, while Vmax increased, which indicates a higher affinity and activity retention by immobilized β-galactosidase. The bound enzyme retained 83% activity even after its 8th successive reuse. The adsorbed enzyme lost only 21% of its initial activity during storage at 4 °C, while the free β-galactosidase retained only 49% activity under similar storage conditions. The genotoxicity assessment revealed that the nanocomposites showed negligible toxicity to pBR322 DNA plasmid and human lymphocytes. In view of its easy production, non-toxic nature, improved stability against various denaturants and excellent reusability, the versatile Gr@Fe3O4 NCs can serve as an ideal support for the immobilization of other enzymes as well, and it may find applications in constructing biosensors and producing lactose-free dairy products to feed lactose intolerant patients.
1. Introduction
Enzymatic hydrolysis of lactose is one of the most important biotechnological processes in the food industry. β-Galactosidase is extensively employed in the hydrolysis of lactose present in milk and whey. This enzyme is widely present in plants, animals and microbes. The main benefit of the hydrolyzed products is to minimize the chances of lactose intolerance. Another advantage is the formation of galactose-oligosaccharides, important probiotic compounds. The lactose-free products thus formed, have improved sweetness, and are easily digested by lactose intolerant people.1,2 Treatment of cheese whey with β-galactosidase increases its fermentability to ethanol, thereby reducing the pollution created by disposal of large volumes of this by-product into the environment.3 However, free enzymes cannot be used continuously in industries because of their instability in harsh environmental conditions, non-reusability and difficult recovery. Immobilized enzymes overcome these limitations by providing reusability, operational and long term stability, as well as continuous processing.4,5 Various methods such as simple adsorption, entrapment, covalent coupling, crosslinking, etc., have been employed for immobilization of β-galactosidase on different supports.6 Immobilization of industrially important enzymes on nanomaterial supports has paved the way for a myriad of application-based commercialization in various fields of science because of the exceptional properties such as large surface area to volume ratio, high catalytic efficiency, high surface reactivity, as well as strong adsorption ability.7,8 As biotechnology deals with nanometer sized molecules like proteins and nucleic acids, the ability to tailor the properties of materials at nanoscale offers excellent prospects for increasing the performance of enzyme-based biosensors.6,9Graphene, an allotrope of carbon (C) comprises of a single layer of carbon atoms arranged in a 2-D honey-combed network.10 Recently, researchers have gained extreme interest in graphene based products because of its unique electrical, mechanical, thermal and optical properties within a single sheet of sp2 bonded C atoms.11 Being an inexpensive material with excellent electronic properties and high surface area, graphene can be used as an ideal support for the immobilization of enzymes and thus finds its use in the construction of biosensors.12,13 Restacking of the graphene sheets to form graphite can be prevented by reacting it with inorganic particles leading to a new class of graphene-based nanocomposites. Recently, among the various nanostructures, magnetic nanoparticles (MNPs) have been brought into sharp focus in the field of enzyme immobilization largely due to their super-paramagnetic properties, low toxicity and biocompatibility in physiological environments.14 Depositing MNPs on graphene will impart an unprecedented combination of magnetic and electrical properties upon graphene making the composite a perfect material to be used in a wide range of applications like drug delivery, biofuel cells, biosensing, catalysis, etc.15 Till date, few efforts have been focussed to use graphene-iron oxide nanocomposites (Gr@Fe3O4 NCs) as an efficient immobilization support for various enzymes.9,16
In this study, we have prepared Gr@Fe3O4 NCs by co-precipitation method, and this support has been employed for the immobilization of β-galactosidase by means of simple adsorption. The Fe3O4 NPs, Gr@Fe3O4 NCs, and the immobilized enzyme were characterized by X-ray diffraction (XRD), vibrating sample magnetometer (VSM), and FT-Raman spectroscopy. The binding of β-galactosidase on Gr@Fe3O4 NCs was confirmed by Fourier transform infra-red spectroscopy (FTIR), transmission electron microscopy (TEM), and scanning electron microscopy (SEM). Effect of pH, temperature and galactose on the activity of free and immobilized enzyme have been compared. Reusability and storage stability of the immobilized enzyme were also monitored. Genotoxicity of the free as well as the enzyme bound support was analyzed with the help of plasmid nicking and comet assay.
2. Materials and methods
2.1. Materials
Aspergillus oryzae β-galactosidase (3.2.1.23), Histopaque-1077, RPMI-1640 medium and low melting agarose were obtained from Sigma-Aldrich Chemicals Co. (USA). o-Nitrophenyl β-D-galactopyranoside (ONPG) was purchased from Sisco Research Laboratories (SRL), Mumbai, India. All other reagents and chemicals were of analytical grade and were used without any further purification.2.2. Synthesis and characterization of Gr@Fe3O4 NCs
Graphite oxide (GO) was synthesised via a modified Hummers method, and was subsequently exfoliated by ultrasonication to attain an aqueous dispersion of GO. The Gr@Fe3O4 NCs were synthesised by co-precipitating pre-hydrolysed ferric and ferrous salts in the presence of GO.17 Briefly, an aqueous solution (100 mL) containing FeCl3·6H2O (4.0 mM) and FeCl2·4H2O (2.0 mM) was prepared with an initial pH of 1.48. However, to overcome the stacking of the GO sheets at low pH, GO was not added into the mixture until the pH was adjusted to pH 4 via addition of NaOH (1.0 M). Subsequently, the GO solution (50 mL, 0.55 mg mL−1) was gradually added into the pH 4 solution and stirred for another 30 min in order to obtain a stable and homogeneous mixture. An appropriate amount of NaOH (1.0 M) was continuously added into the mixture until the pH reached 10. The mixture was then constantly stirred for another 30 min at room temperature. The resulting black precipitate was magnetically separated and washed thrice with deionized water and ethanol and finally dried in an oven at 60 °C for 48 h. Pure Fe3O4 NPs were also prepared via an analogous method without the addition of GO solution.The sizes and morphologies of Gr@Fe3O4 NCs, Fe3O4 NPs and adsorbed β-galactosidase were examined by TEM and SEM, respectively. Small amount of samples from Fe3O4 NPs, Gr@Fe3O4 NCs, and bound NCs were prepared for TEM analysis by drop coating diluted solutions on carbon coated copper grids at normal atmospheric conditions. The obtained samples were then analysed under JEOL-2100 transmission electron microscope at 200 kV. Small samples from Gr@Fe3O4 NCs, Fe3O4 NPs, and air dried immobilized enzyme were mounted on carbon tape on a copper stub, followed by 60 nm of gold deposition using a sputter coater. The samples were examined under JSM-6510 LV scanning electron microscope at 15 kV. The crystal structures of the NPs and the NCs were interpreted by XRD pattern obtained by X-ray diffractometer (Rigaku, Japan-Miniflex-II) using a monochromatized X-ray beam with nickel-filtered Cu Kα radiation. The scan speed of 4 steps per s with an angle range (20−80°) was used. The Raman spectra of Fe3O4 NPs and Gr@Fe3O4 NCs were recorded on a BRUKER RFS 27: Stand alone FT-Raman Spectrometer using laser source Nd:YAG 1064 nm and a resolution of 2 cm−1. FTIR spectra of the Fe3O4 NPs, Gr@Fe3O4 NCs and air dried immobilized enzyme were recorded using FTIR-Perkin Elmer instrument, USA (Spectrum-II, wave no. 4000-400). The calibration was done by polystyrene film. Phase KBr pellet method was employed to run the samples. Background spectra of a clean atmosphere were acquired prior to each sample measurement using the identical acquisition parameters. The magnetic properties of Fe3O4 NPs as well as Gr@Fe3O4 NCs were characterized with a vibrating sample magnetometer (VSM, ADE EV5) by measuring the applied field dependence of magnetization between −15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 15000H (g) at room temperature. The enzyme concentration was measured by UV-vis spectrophotometer (Shimadzu, UV-1800).
000 and 15000H (g) at room temperature. The enzyme concentration was measured by UV-vis spectrophotometer (Shimadzu, UV-1800).
2.3. Immobilization of β-galactosidase on Gr@Fe3O4 NCs
Free β-galactosidase was independently adsorbed on varying concentrations (ranging from 2–18 mg) of Gr@Fe3O4 NCs by continuously shaking the mixture in sodium acetate buffer, pH 4.5, at 25 °C. The enzyme concentration was also varied from 1 mg mL−1 to 5 mg mL−1, keeping the NCs concentration (10 mg) fixed. The immobilization time was increased from 1 h to 10 h.Gr@Fe3O4 NCs (500 mg) were suspended and sonicated in 0.1 M sodium acetate buffer, pH 4.5. β-Galactosidase (49.12 U) was then loaded in colloidal suspension. The mixture was gently stirred for 5 h and then either magnetically separated or centrifuged at 3000 × g for 10 min at 4 °C. The precipitate obtained was washed thrice with assay buffer and preserved at 4 °C for further use. The supernatant and all the washes were also stored at 4 °C for the calculation of immobilization yield.7
2.4. Stability and kinetic studies of the immobilized β-galactosidase
Free and adsorbed β-galactosidase was stored at 4 °C in 0.1 M sodium acetate buffer, pH 4.5 for 2 months. Aliquots from each preparation (50 μL) were taken in triplicates at a gap of 5 days and were then analyzed for the activity. The activity determined on the first day was taken as control (100%) for the calculation of remaining percent activity.18
2.5. Genotoxicity assessment of Gr@Fe3O4 NCs
Plasmid nicking assay was performed according to the procedure described by Khan et al.8 Reaction mixture (30 μL) containing 10 mM Tris–HCl buffer (pH 7.5), pBR322 DNA plasmid (0.5 μg) with free enzyme (0.08 U), Gr@Fe3O4 NCs (40 μg), and immobilized enzyme (10 μL) were incubated for 2 h at 37 °C. Ten microlitre of a solution containing 40 mM EDTA, 0.05% (w/v) bromophenol blue (tracking dye), and 50% (v/v) glycerol was added after incubation and the solution was then subjected to electrophoresis in submarine 1.0% (w/v) agarose gel. Ethidium bromide stained gel was then viewed and photographed on a UV-transilluminator.Comet assay was done according to the procedure described by Khan et al.20 Fully frosted microscopic slides pre-coated with 1.0% (w/v) normal melting agarose (PBS buffer lacking Ca2+ and Mg2+ ions) were used at 50 °C. Diluted lymphocytes (10000 cells) were mixed with 80 μL of 1.0% (w/v) low melting point agarose to form a cell suspension and were pipetted over the first layer and covered immediately by a cover slip. The slides were put on a flat tray and maintained on ice for 10 min to solidify agarose. Cover slips were removed and 0.5% (w/v) low melting point agarose (80 μL) was layered which was then allowed to solidify on ice for 5 min. Within 20 min of immediately removing the cover slips, cells were individually treated with free enzyme (0.08 U), free NCs (200 μg) and adsorbed β-galactosidase (50 μL) for 1 h at 4 °C. All the reactions on the slide were done in triplicates. The slides were immersed in cold lysis buffer containing 2.5 M NaCl, 100 mM EDTA and 10 mM Tris–HCl, pH 10 for 1 h at 4 °C and 1% Triton-X 100 was added (just prior to use). After lysis, DNA was allowed to unwind for 30 min in an alkaline electrophoretic solution consisting of 300 mM NaOH and 1.0 mM EDTA, pH > 13. Electrophoresis was run at 4 °C in field strength of 0.7 V cm−1 and 300 mA current. The slides were neutralized with cold buffer of 0.4 M Tris and pH 7.5 and subsequently stained with 75 mL ethidium bromide (20 mg mL−1) and covered with glass slips. These were then placed in a humidified chamber to prevent drying of gel and were analyzed the same day. Slides were scored using an image analysis system (Comet 5.5, Kinetic Imaging, Liverpool, UK) attached to an Olympus fluorescent microscope (CX 41, Olympus Optical Co., Tokyo, Japan) and a COHU 4910-integrated CC camera (equipped with 510–560 nm excitation and 590 nm barrier filters) (COHU, San Diego, CA, USA). Comets were scored at 100× magnification. Images from 50 cells (25 from each replicate slide) were analyzed. Tail length in μm (migration of DNA from the nucleus) was the parameter taken to assess DNA damage to human lymphocytes and it was automatically generated by the Comet 5.5 image analysis system.
2.6. Assay of β-galactosidase
The activity of free and bound β-galactosidase was determined by measuring the release of o-nitrophenol from ONPG at 405 nm.6 The reaction was performed by continuous shaking in an assay volume of 2.0 mL containing 1.60 mL and 1.65 mL of 0.1 M sodium acetate buffer (for free and immobilized enzyme, respectively) of pH 4.5, 0.08 U β-galactosidase (free-100 μL, immobilized-50 μL) and 0.3 mL of 20 mM ONPG at 37 °C for 15 min. The reaction was terminated by adding 2.0 mL of 1.0 M sodium carbonate solution, and product formation was spectrophotometrically measured at 405 nm.One unit (1.0 U) of β-galactosidase activity is defined as the amount of enzyme that catalyzes the release of 1.0 μmol of o-nitrophenol (εm = 4500 L mol−1 cm−1 min−1) under the specified assay conditions.
2.7. Ethical statement
Five millilitre of fresh human blood (self donor) was taken for the genotoxic assessment of the immobilization support in ‘comet assay’. According to the ethical guidelines of Indian Council for Medical Research, New Delhi, India, Chapter-II, page no. 11-12, ethical approval for this research work was not deemed to be necessary. According to their guidelines, proposals which present less than minimal risks are exempted from the ethical review process.2.8. Estimation of protein
Protein concentration was determined according to the procedure described by Lowry et al.21 Bovine serum albumin was used as a standard protein.2.9. Statistical analysis
Each value represents the mean for three independent experiments, and statistical analysis for comparison between free and immobilized β-galactosidase was performed by one way ANOVA test. The data expressed in various studies were plotted using origin 6.1 and expressed with standard deviation of error. Statistical significance was measured at p-values ≤ 0.05.3. Results and discussion
3.1. Synthesis and characterization
The synthetic methods for the preparation of Gr@Fe3O4 NCs usually require tedious steps and toxic reagents such as hydrazine,22 and NaBH4,23 till now, only a few efforts have been made to construct an easy and eco-friendly method for the synthesis of Gr@Fe3O4 NCs.24,25 Hence, there is an urgent need to develop a safe and efficient method to prepare Gr@Fe3O4 NCs. Since pure graphene sheet has rare functional moieties on its surface, GO has proved to be a suitable substitute for graphene on account of having a large number of chemically reactive sites, such as the epoxy and the hydroxyl groups on the basal plane and the carboxylic groups at the surface (Fig. 1), and due to presence of the π–π conjugated system, which could oxidize Fe2+ ions for the nucleation and growth of Fe3O4 nanoparticles.9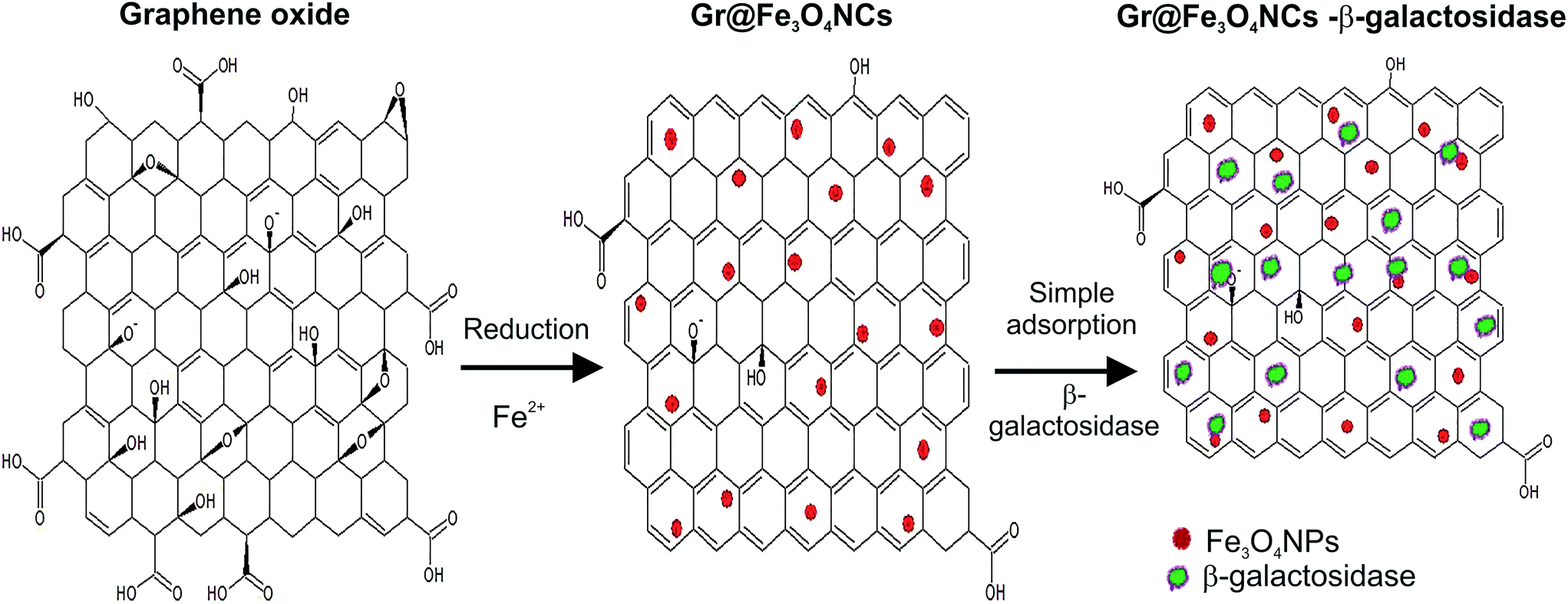 | ||
| Fig. 1 Schematic illustration of the method of preparation of Gr@Fe3O4 NCs and immobilization of β-galactosidase. | ||
Fig. 1 depicts the procedure of preparation of Gr@Fe3O4 NCs and immobilization of β-galactosidase. GO is reduced upon addition of Fe2+ ions which attach to the surface of GO through electrostatic interactions and are oxidized to Fe3+ by the O2− groups present on the surfaces of GO. To prevent restacking of GO sheets, the pH value is increased via addition of NaOH. Consequently, the Fe2+ ions co-precipitate with the acquired Fe3+ ions into Fe3O4, leading to the formation of Gr@Fe3O4 NCs.9 The enzyme is then directly adsorbed on the surface of the obtained NCs via hydrogen bonding and van der Waals interactions between the protein and the Gr@Fe3O4 NCs surface.26
Free and enzyme bound functionalized graphene (Gr@Fe3O4 NCs) were characterized by TEM, SEM, and FTIR spectroscopy. XRD and VSM were done to monitor the crystallinity and to confirm the magnetic character of the NPs as well as the NCs. The particle sizes and surface morphologies of pure Fe3O4 NPs, Gr@Fe3O4 NCs, and immobilized β-galactosidase were determined by TEM and SEM. The incorporation of Fe3O4 NPs onto graphene sheets was confirmed by TEM and SEM images as illustrated in Fig. 2(a–d). As seen in the representative TEM images of Gr@Fe3O4. The surface of graphene is densely covered by cylindrical shaped Fe3O4 NPs with an average size of about 25 nm (Fig. 2(c and d)). The distribution of Fe3O4 NPs on graphene sheet is even, and no aggregation or large vacancy on graphene was found. Selection area electron diffraction (SAED) pattern shows that the NCs are polycrystalline with a slight visibility of circular rings (Fig. 2(b)). He and Gao15 also reported similar findings. Fig. 2(e–h) demonstrates the TEM and SEM images of Fe3O4 NPs. Roughly spherical Fe3O4 NPs were observed with an average particle size of 55 nm. SAED pattern indicates that Fe3O4 NPs were crystalline in nature (Fig. 2(f)). The space resolved lattice fringes as shown in Fig. 2(h) agree well with the lattice spacing planes of Fe3O4 NPs. Similar results have also been reported earlier.17 Binding of β-galactosidase to the immobilization matrix was confirmed by TEM and SEM images (Fig. 3). The TEM analysis indicates adsorption of protein onto functionalized graphene NCs (Fig. 3(a–f)). Rigid binding of enzyme to the immobilization support can be clearly illustrated by SEM analysis at 50 μm, 10 μm and 5 μm (Fig. 3(d–f)). Moreover, the immobilization is also responsible for alteration in the characteristic SAED pattern. Fig. 3(c) shows distinguishable concentric rings which indicated that the Gr@Fe3O4 NCs were present in highly crystalline form after binding to β-galactosidase.
 | ||
| Fig. 2 (a) SEM image of Gr@Fe3O4 NCs, (b) SAED pattern of Gr@Fe3O4 NCs, (c & d) TEM images of Gr@Fe3O4 NCs, (e) SEM image of Fe3O4 NPs (f) SAED pattern of Fe3O4 NPs (g) TEM image of Fe3O4 NPs, (h) lattice fringes of a single Fe3O4 NPs. | ||
 | ||
| Fig. 3 TEM and SEM images of enzyme bound Gr@Fe3O4 NCs (a & b) TEM images (c) SAED pattern (d–f) SEM images at 50 μm, 10 μm and 5 μm. | ||
Fig. 4(a) demonstrates the XRD patterns of both the Fe3O4 NPs and the Gr@Fe3O4 NCs. The crystallinity in both the samples was corroborated by peak positions at 2θ = 32.31°, 34.65°, 39.92°, 48.71°, and 53.26° for Fe3O4 NPs and 32.49°, 34.99°, 40.14°, 48.88°, and 53.57° for Gr@Fe3O4 NCs, respectively. These peaks were consistent with the standard XRD data in JCDPS (file no. 19-0629). No diffraction peaks of other impurities were detected, confirming that both samples prepared were pure and crystalline in nature which is in accordance with earlier published work.9 Raman spectroscopic studies were also performed in order to assess the incorporation of Fe3O4 NPs on the graphene sheets. Fig. 4(b) shows the Raman spectra of Fe3O4 NPs and Gr@Fe3O4 NCs. The Fe3O4 NPs spectrum showed a major peak at 2907 cm−1 and a small peak at 1382 cm−1, corresponding to C–H and CH3 bonding, respectively. The Raman spectrum of Gr@Fe3O4 NCs reveals two prominent peaks at 1602 cm−1 and 1310 cm−1 which confirms the presence of well documented D (defects/disorder-induced modes) and G bands (in-plane stretching tangential modes) in graphene sheets. The ID/IG ratio for GO is normally more than 1.0, as has been well acknowledged by researchers,27 but on reduction of GO to graphene in the presence of Fe2+ ions, the intensity ID/IG ratio decreased to 0.82. Similar findings have also been previously discussed.28 These bands were not observed in Fe3O4 NPs. The peak at 1602 cm−1 further signifies the presence of aromatic ring chain vibrations (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and denotes attachment of Fe3O4 NPs to graphene sheets.
C) and denotes attachment of Fe3O4 NPs to graphene sheets.
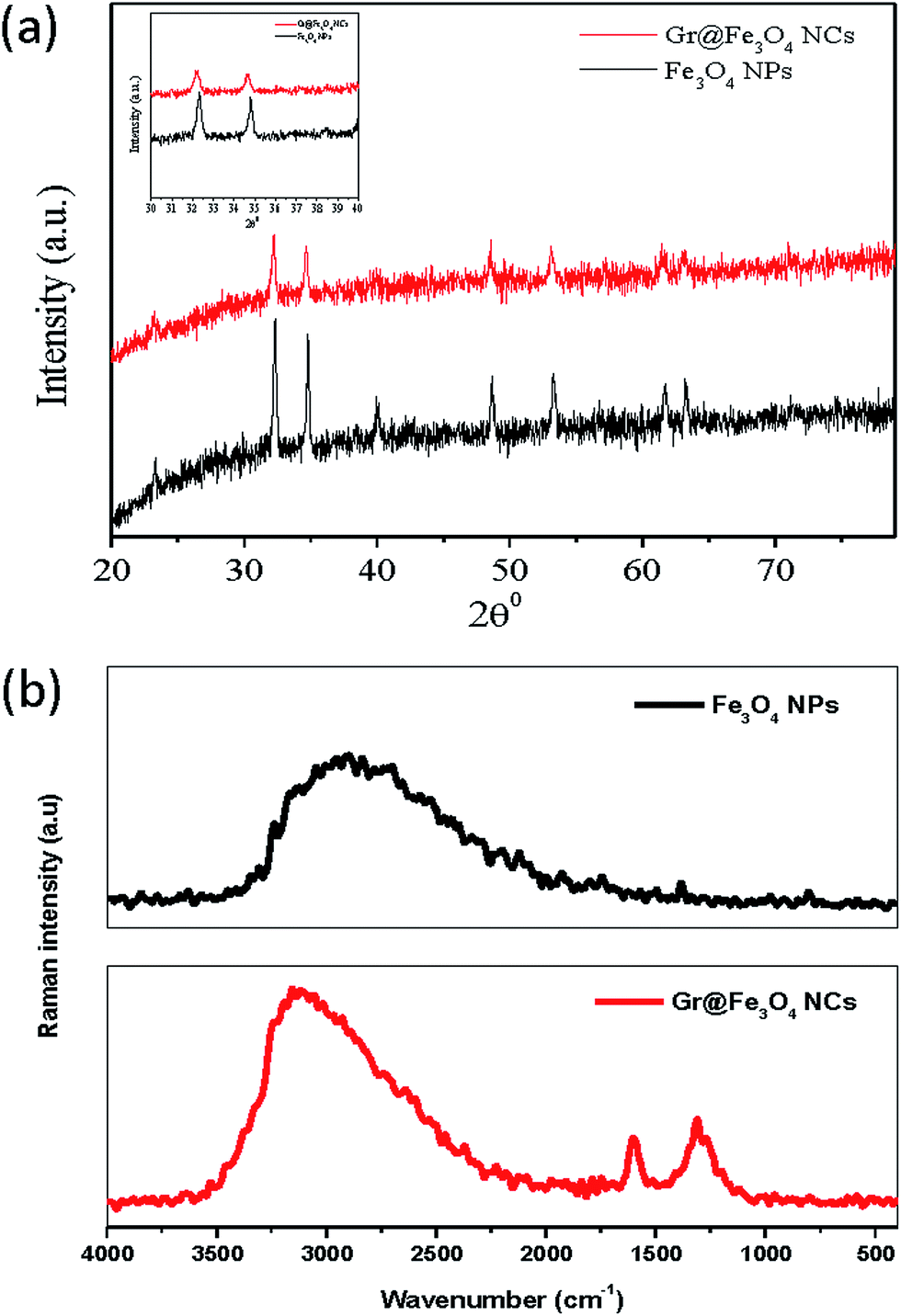 | ||
| Fig. 4 (a) XRD pattern of Gr@Fe3O4 NCs and Fe3O4 NPs, (b) FT-Raman spectra of Fe3O4 NPs and Gr@Fe3O4 NCs. | ||
To assess the magnetic properties of the Fe3O4 NPs as well as the Gr@Fe3O4 NCs, these were exposed to external magnetic field sweeping from −15000 to 15000H (g) at room temperature by the use of vibrating sample magnetometer. The magnetic hysteresis (M–H) loops of Gr@Fe3O4 NCs exhibited an S-shaped curve with negligible retentivity and coercivity (Fig. 5(a)), indicating no remaining magnetization upon removal of external magnetic field.15 The NCs hence showed super-paramagnetic behaviour while the naked Fe3O4 NPs exhibited paramagnetic behaviour. The saturation magnetization (Ms) for Gr@Fe3O4 NCs was found to be about two folds greater than that of the naked ones indicating that the particles are highly super-paramagnetic which is a characteristic of a ferromagnetic material.
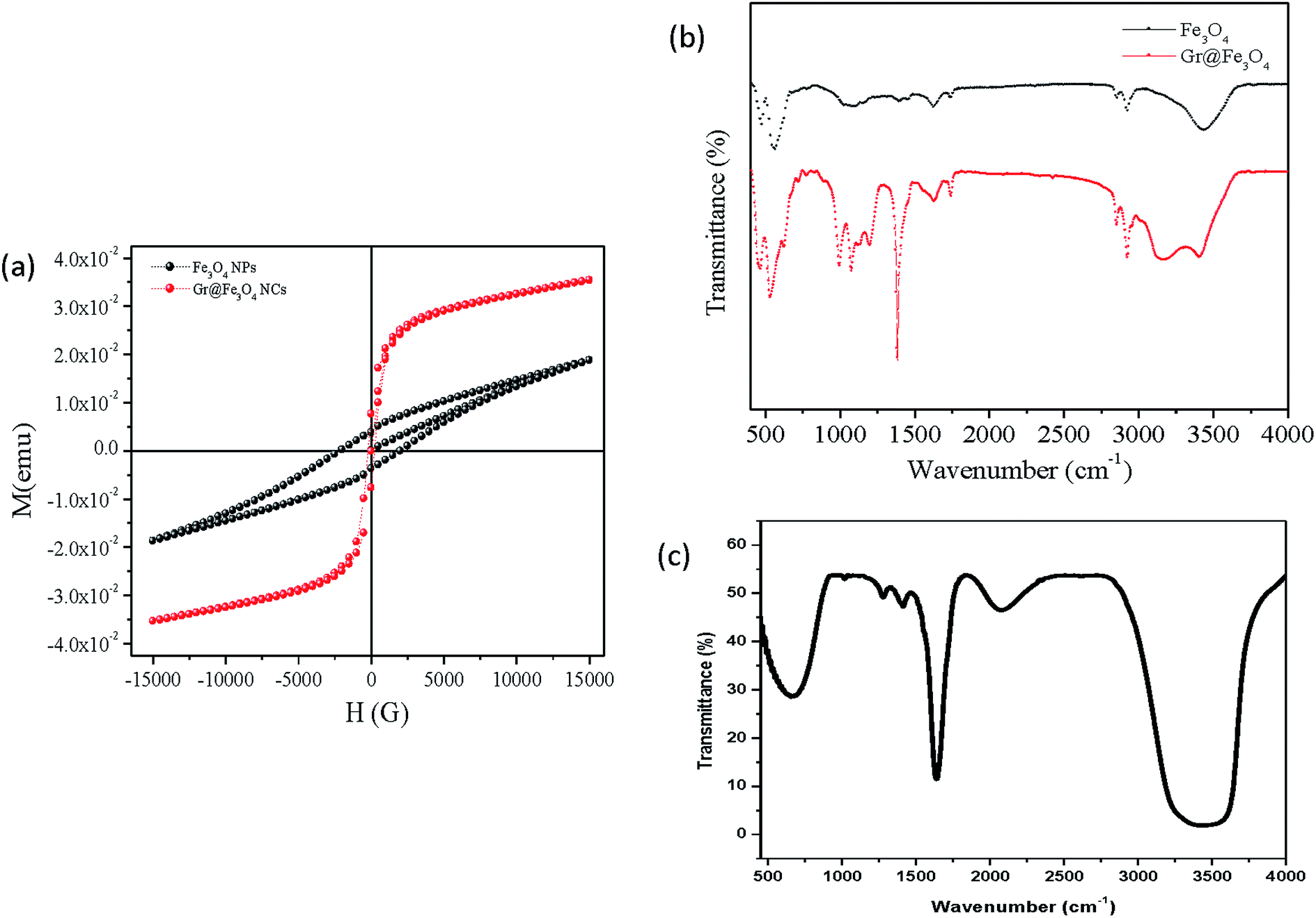 | ||
| Fig. 5 (a) Magnetic hysteresis loops of Fe3O4 NPs and Gr@Fe3O4 NCs measured at room temperature, (b) FTIR spectra of Fe3O4 NPs and Gr@Fe3O4 NCs and (c) enzyme bound Gr@Fe3O4 NCs. | ||
Fig. 5(b) demonstrates the characteristic FTIR spectra of Fe3O4 NPs and of free as well as bound Gr@Fe3O4 NCs. In the first two samples, the peaks at 568 cm−1 and 534 cm−1, respectively, correspond to the Fe–O vibrations related to the magnetite phase.29 The peak at 1631 cm−1 in the absorption spectra of Gr@Fe3O4 NCs is the characteristic peak for CC stretch indicating the presence of graphene. The peaks observed at 2925 cm−1 and 2854 cm−1 in Fe3O4 NPs and Gr@Fe3O4 NCs, respectively, are due to C–H stretching. The FTIR bands corresponding to the OH stretching mode are shifted to the lower wave number in the FTIR spectra of the Gr@Fe3O4 NCs, 3407 cm−1 compared to the pure Fe3O4 NPs, 3435 cm−1 (Fig. 5(b)) due to the interaction taking place between NPs and the graphene sheets.13 This signifies the role of O–H group during the interaction of NPs and graphene. The absorption spectrum of the immobilized enzyme confirms the binding of β-galactosidase to the Gr@Fe3O4 NCs (Fig. 5(c)). The stretching of carbonyl group of β-galactosidase was observed by broadening of peaks from 3320 cm−1 and 3547 cm−1 which ascertains the binding of enzyme to the support.30 Furthermore, the peak at 1636 cm−1 corresponding to N–H bend also confirms the presence of amide band I (Fig. 5(c)).
3.2. Binding studies of immobilized β-galactosidase
Although the preparation of carrier is quite important for the immobilization process as well as the performance of immobilized enzyme, the selection and optimization of immobilization conditions is also critical for immobilized enzyme to exhibit its maximal activity. Therefore, three parameters affecting the immobilization of β-galactosidase were investigated in the present study. In order to investigate the effect of concentration of NCs on the immobilization yield, the magnetic graphene NCs with different concentrations ranging from 2 mg to 18 mg of dry support was used keeping other parameters fixed (enzyme concentration: 1 mg mL−1, immobilization time, 5 h). Initially, the immobilization yield increased with the increase of Gr@Fe3O4 NCs concentration and maximum immobilization yield was 90% at 10 mg of protein. Further increase in NCs concentration showed no significant effect on the immobilization yield due to the fact that the protein fully binds to the support at this concentration and reaches at saturation level.31Effect of different enzyme concentrations on enzyme immobilization was also examined. The concentrations used varied (1–5 mg mL−1) keeping other conditions like concentration of support (10 mg dry weight) and immobilization time (5 h) constant. The maximum immobilization yield, 90% was obtained at initial enzyme concentration of 1 mg mL−1 after which it decreased gradually from 2–5 mg mL−1. This phenomenon can be attributed to diffusion limitation theory. At high enzyme concentrations, the neighbouring proteins block access of substrate to the active sites.32 To determine the optimum immobilized time, immobilization was conducted at varied times with other parameters fixed (1 mg mL−1 enzyme and 10 mg Gr@Fe3O4 NCs). The immobilization time had a significant influence on the yield of immobilization in the first 5 h. The enzyme loading reached 90% at the end of 5 h, and then remained nearly constant with prolonged incubation. This result was in accordance with that earlier reported which also proved the immobilization process as swift.33
Table S1† demonstrates the calculation of immobilization yield of β-galactosidase on Gr@Fe3O4 NCs in terms of enzyme units (U). Consequently, the immobilization strategy achieved 89% efficiency and 90% yield after optimizing all the above factors. The immobilization efficiency is defined as the ratio of the activity of the immobilized enzyme to the activity of the free enzyme used.
3.3. Stability and kinetic studies of immobilized β-galactosidase
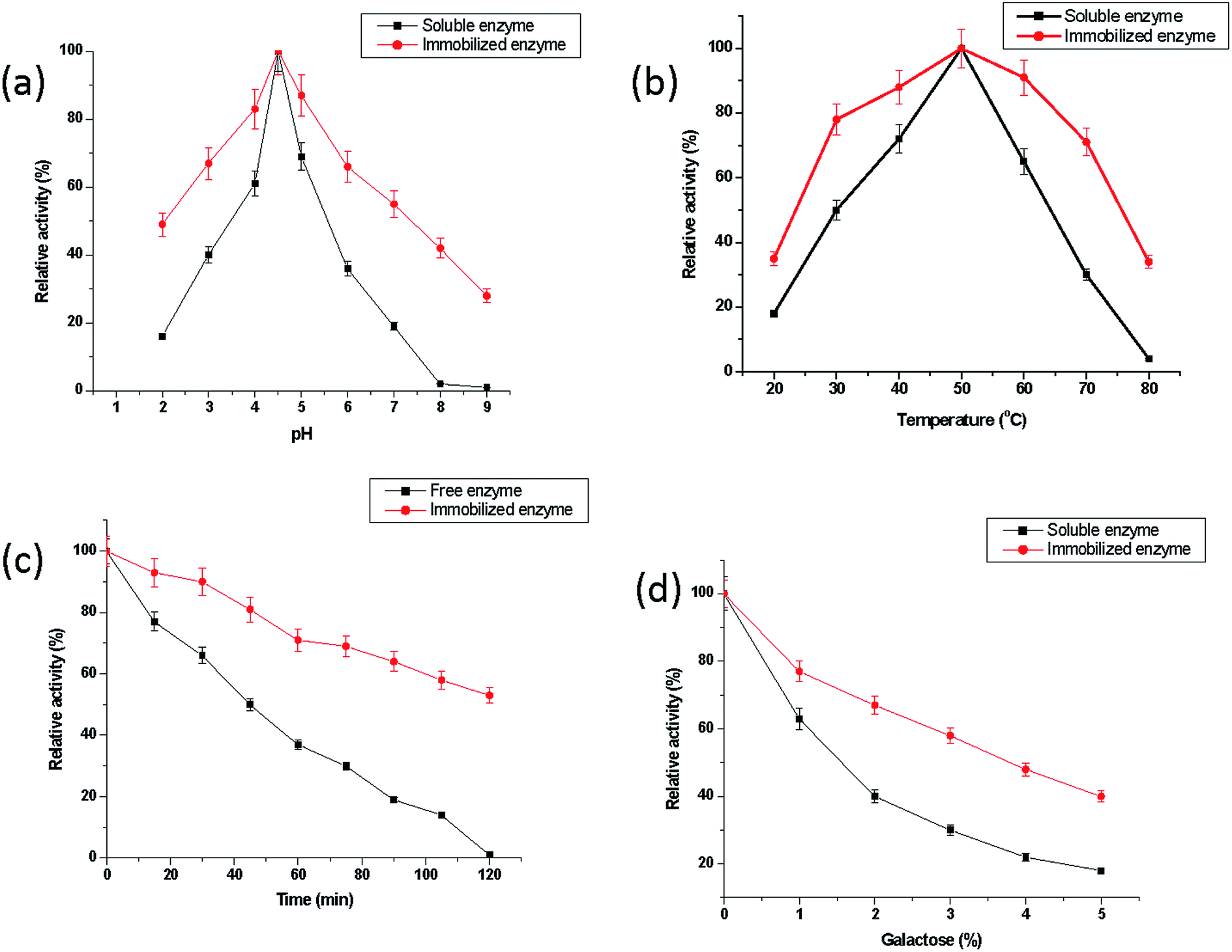 | ||
| Fig. 6 (a) pH-Activity profiles for free and immobilized β-galactosidase. The activity of free and immobilized β-galactosidase (0.08 U) was measured in the buffers of varying pH (2.0–9.0). The buffers used were glycine–HCl (pH 2.0 & 3.0), sodium acetate (pH 4.0 & 5.0), sodium phosphate (6.0 & 7.0) and Tris–HCl (pH 8.0 & 9.0). Molarity for each buffer used was 0.1 M. The activity at pH 4.5 was considered as control (100%) for the calculation of residual percent activity, (b) temperature-activity profiles for free and immobilized β-galactosidase. The activity of free and immobilized galactosidase (0.08 U) was measured in 0.1 M sodium acetate buffer, pH 4.5 at different temperatures (20–80 °C) for 15 min. The activity obtained at 50 °C was considered as control (100%) for the calculation of residual percent activity, (c) thermal denaturation of free and immobilized β-galactosidase. Free and immobilized galactosidase (0.08 U) were incubated at 60 °C in 0.1 M sodium acetate buffer, pH 4.5 for 2 h. Aliquots of each preparation (20 μL) were taken out at an interval of every 15 min and chilled quickly in crushed ice for 5 min. The activity of the enzyme obtained without incubation at 60 °C was taken as control (100%) for the calculation of residual percent activity, (d) effect of galactose on the activity of free and immobilized β-galactosidase. The activity of free and immobilized galactosidase (0.08 U) preparations was measured in the presence of increasing concentrations of galactose (1.0–5.0%, w/v) in 0.1 M sodium acetate buffer, pH 4.5 for 1 h at 37 °C. The activity of the enzyme in the absence of galactose was considered as control (100%) for the calculation of residual percent activity at different concentrations of galactose. Data represents mean ± S.D of three individual experiments performed in duplicates, p-value ≤ 0.05 for each figure. | ||
Fig. 6(b) illustrates the temperature-activity profiles for free and bound enzyme. The temperature-optima for both free and immobilized β-galactosidase were observed at 50 °C. The immobilized enzyme again exhibited a broadening in the temperature-activity from 30 °C to 70 °C as compared to its free form. It was observed that Gr@Fe3O4 NCs bound β-galactosidase exhibited 71% of the maximum activity at 70 °C while free enzyme showed only 31% activity under identical experimental conditions. Similar findings were also reported by Husain et al.7 In this case, a broadening in temperature-optima was observed from 50 °C to 60 °C.
The thermal stability of the free and immobilized enzyme has been compared in Fig. 6(c). It showed that free β-galactosidase enzyme lost about 99% activity after 2 h incubation at 60 °C, while Gr@Fe3O4 NCs bound β-galactosidase exhibited 53% activity under identical incubation conditions. Husain et al.7 have also affirmed greater thermal stability exhibited by the ZnO bound β-galactosidase as compared to its free form under same experimental conditions. The loss in enzyme activity at higher temperatures can be attributed to denaturation of enzyme molecules resulting in the rupturing of polypeptide chain and degradation of the polymer matrix. However, the immobilization matrix imparts rigidity to the enzyme, thereby making the immobilized enzyme more resistant against thermal denaturation.19
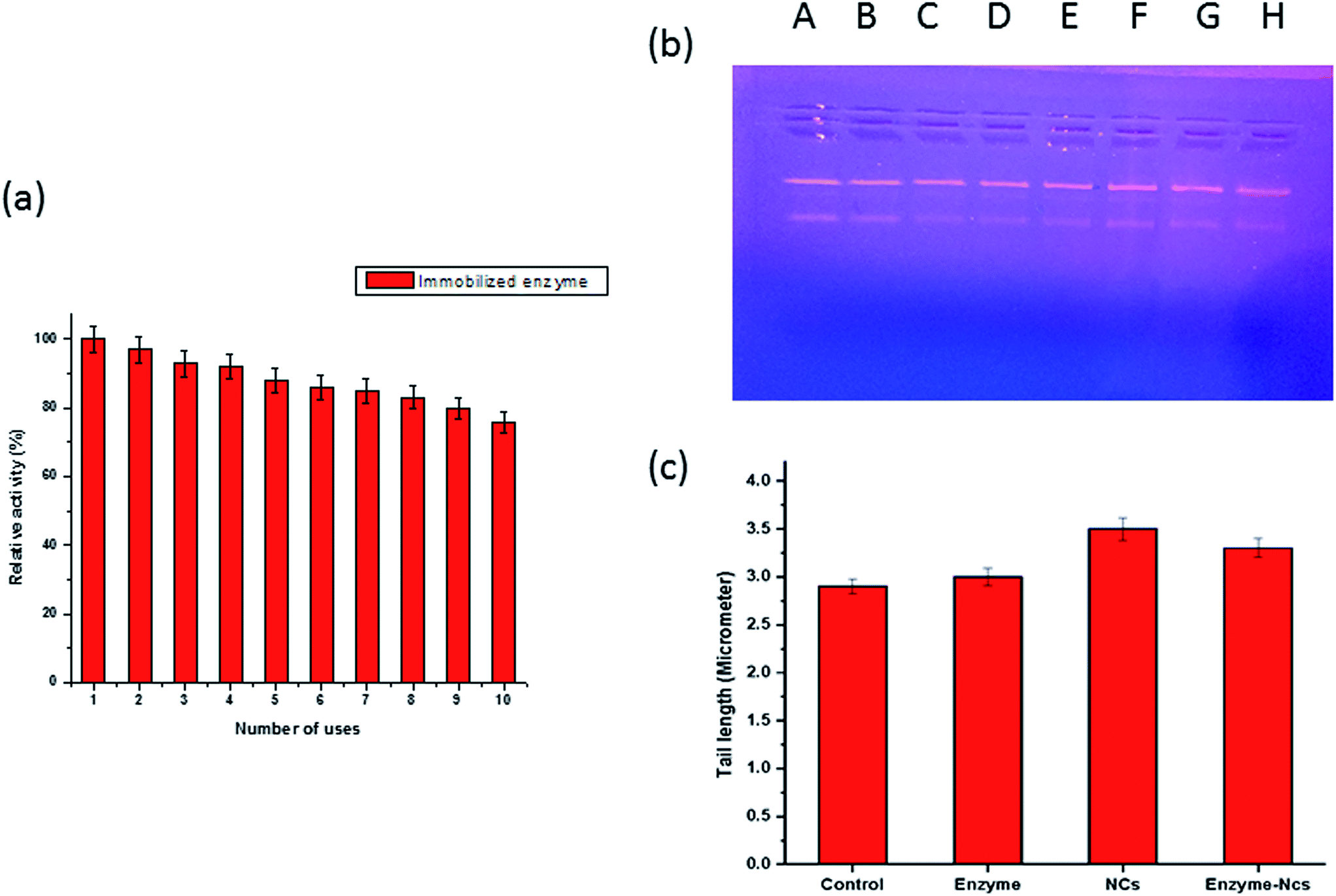 | ||
| Fig. 7 (a) Reusability assay of immobilized β-galactosidase. The reusability of adsorbed β-galactosidase was monitored for 10 successive days. The aliquots from preparation were taken in triplicates and the activity determined on the first day was taken as control (100%) for the calculation of residual activity after each use. p-Value ≤ 0.05 was considered statistically significant, (b) plasmid nicking assay: Lane A, B: pBR322 DNA alone, Lane C & D: pBR322 DNA + enzyme (0.08 U), Lane E & F: pBR322 DNA + Gr@Fe3O4 NCs (40 μg), Lane G & H: pBR322 DNA + Gr@Fe3O4 NCs bound β-galactosidase, (c) DNA damage in human lymphocytes by enzyme alone (0.08 U), Gr@Fe3O4 NCs (200 μg) and Gr@Fe3O4 NCs-bound enzyme. Data represents mean ± S.D of three individual experiments performed in triplicates, p-value ≤ 0.05. | ||
| Number of days | Remaining activity (%) | |
|---|---|---|
| Free β-galactosidase | Immobilized β-galactosidase | |
| Control | 100 | 100 |
| 5 | 94 ± 4.0 | 99 ± 5.0 |
| 10 | 87 ± 3.8 | 97 ± 4.8 |
| 15 | 84 ± 3.7 | 94 ± 4.7 |
| 20 | 76 ± 3.7 | 93 ± 4.6 |
| 25 | 72 ± 3.6 | 92 ± 4.6 |
| 30 | 67 ± 3.4 | 89 ± 4.4 |
| 35 | 63 ± 3.3 | 87 ± 4.3 |
| 40 | 62 ± 3.3 | 85 ± 4.3 |
| 45 | 60 ± 3.2 | 84 ± 4.2 |
| 50 | 56 ± 3.2 | 83 ± 4.0 |
| 55 | 52 ± 3.1 | 81 ± 4.0 |
| 60 | 49 ± 3.0 | 79 ± 3.8 |
3.4. Toxicity assessment
It was essential to demonstrate the genotoxic effect of Gr@Fe3O4 NCs so that it could be used as an immobilization matrix for β-galactosidase in biosensor applications and most importantly in the food industry. The plasmid nicking assay is one of the important parameters to study for analyzing the effect of various genotoxic chemicals on the integrity of DNA. In our study, pBR322 DNA plasmid was exposed to the enzyme alone, the Gr@Fe3O4 NCs and bound β-galactosidase. As evident from Fig. 7(b), the damaging effect of Gr@Fe3O4 NCs on the plasmid DNA was found to be negligible since the band intensity of the treated sample was similar to that of the control in all the cases. Similar findings have also been reported earlier.30Single cell gel electrophoresis, commonly known as comet assay is a very sensitive technique to assess DNA strand breaks in individual cells and has been recommended by various workers as a valid indicator of genotoxicity.40,41 The DNA of human lymphocytes were treated in vitro to enzyme alone, the Gr@Fe3O4 NCs, as well as adsorbed β-galactosidase, and the tail length, which is a parameter of DNA damage was plotted against concentration of different samples. Fig. 7(c) depicts insignificant DNA damage in the lymphocytes when exposed to the Gr@Fe3O4 NCs alone as well as the bound form with tail length of around 3.5 μm and 3.3 μm, respectively, as compared to control (2.9 μm), clearly suggesting that the support which we have used for immobilization of β-galactosidase is non toxic and biocompatible in nature. The toxicity assessment for any immobilized β-galactosidase system is rarely reported till date, though it is used in food industry to feed lactose intolerant people worldwide.30
4. Conclusion
In this study, we have demonstrated highly efficient immobilization of Aspergillus oryzae β-galactosidases onto functionalized graphene sheets (Gr@Fe3O4 NCs). The selection of the support for immobilization revealed a stable and non toxic enzyme–matrix interaction. The thermal and storage stability was greatly improved upon immobilization and the adsorbed enzyme showed greater resistance against galactose, a competitive inhibitor, as compared to its free form. Also, the novel biocatalyst material can be easily separated from the reaction mixture by magnetization or low speed centrifugation, and exhibits excellent reuse characteristics over several successive repeated uses. Hence, this type of robust nanomatrix could serve as a powerful biorecognition probe in biosensor applications for this as well as other enzyme systems. Most importantly, in a country like India where there is high adulteration in milk and its products, we can detect the lactose concentrations in milk easily and efficiently by using this kind of a biosensor. Utility of this method can be further extended in the development of therapeutic agents like targeted drug delivery for lactose intolerant patients. Our findings have paved the way for extensive development of graphene-based nanocomposites with fascinating functionalities in the field of biotechnology and biosensors applications.Conflict of interest
The authors declare that there is no conflict of interest.Acknowledgements
MK gratefully acknowledges the financial assistance in terms of ‘SRF’ by ‘UGC-MANF, New Delhi’. The authors are extremely thankful to Mohd. Shoeb and Shumaila Afrin for providing help in pictorial representation of the data obtained and drawing chemical structures for the manuscript, respectively. USIF, AMU, is duly acknowledged for providing the facilities of TEM and SEM. VSM and FT-Raman technique were performed in SAIF, IIT, Madras.References
- T. Haider and Q. Husain, Biochem. Eng. J., 2009, 43, 307 CrossRef CAS.
- S. A. Ansari and Q. Husain, Pol. J. Chem. Technol., 2011, 13, 15 Search PubMed.
- P. S. Panesar, S. Kumari and R. Panesar, Enzyme Res., 2010, 2010 DOI:10.4061/2010/473137.
- T. Haider and Q. Husain, Int. Dairy J., 2009, 19, 172 CrossRef CAS.
- M. J. Khan, Q. Husain and A. Azam, Biotechnol. Bioprocess Eng., 2012, 17, 377 CrossRef CAS.
- S. A. Ansari and Q. Husain, Biotechnol. Adv., 2012, 30, 512 CrossRef CAS PubMed.
- Q. Husain, S. A. Ansari, F. Alam and A. Azam, Int. J. Biol. Macromol., 2011, 49, 37 CrossRef CAS PubMed.
- M. J. Khan, Q. Husain and S. A. Ansari, Appl. Microbiol. Biotechnol., 2013, 97, 1513 CrossRef CAS PubMed.
- D. Yang, X. Wang, J. Shi, X. Wang, S. Zhang, P. Han and Z. Jiang, Biochem. Eng. J., 2016, 105, 273 CrossRef CAS.
- G. Srivastava, K. Singh, M. Talat, O. N. Srivastava and A. M. Kayastha, PLoS One, 2014, 9 DOI:10.1371/journal.pone.0113408.
- I. V. Pavlidis, M. Patila, U. T. Bomscheuer, D. Gournis and H. Stamatis, Trends Biotechnol., 2014, 32, 312 CrossRef CAS PubMed.
- A. Karimi, A. A. Othman, A. Uzunoglu and S. Andreescu, Nanoscale, 2015, 7, 6909 RSC.
- T. T. Baby, J. Aravind, T. Arackiadoss, R. B. Rakhi and S. Ramaprabhu, Sens. Actuators, B, 2010, 145, 71 CrossRef CAS.
- K. Atacana and M. Ozacar, Colloids Surf., B, 2015, 128, 227 CrossRef PubMed.
- H. He and C. Gao, ACS Appl. Mater. Interfaces, 2010, 2, 3201 CAS.
- B. Jiang, K. Yang, Q. Zhao, Q. Wua, Z. Liang, L. Zhanga, X. Peng and Y. Zhang, J. Chromatogr. A, 2012, 1254, 8 CrossRef CAS PubMed.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang, C. Joao and D. Costa, Sci. Rep., 2014, 4, 4594, DOI:10.1038/srep04594.
- S. A. Ansari and Q. Husain, J. Mol. Catal. B: Enzym., 2010, 63, 68 CrossRef CAS.
- N. Singh, G. Srivastava, M. Talat, H. Raghubhanshi, O. N. Srivastava and A. M. Kayastha, Food Chem., 2014, 142, 430 CrossRef CAS PubMed.
- M. Khan, A. H. Naqvi and M. Ahmad, Toxicol. Rep., 2015, 2, 765 CrossRef CAS.
- O. H. Lowry, N. J. Rosenbrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265 CAS.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979 CrossRef CAS PubMed.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jing, H. K. Jeong, J. M. Kim and J. Y. Choi, Adv. Funct. Mater., 2009, 19, 1987 CrossRef CAS.
- S. Bai, X. Shen, X. Zhong, Y. Liu, G. Zhu, X. Xu and K. Chen, Carbon, 2012, 50, 2337 CrossRef CAS.
- G. Xie, P. Xi, H. Liu, F. Chen, L. Huang, Y. Shi, F. Hou, Z. Zeng, C. Shao and J. Wang, J. Mater. Chem., 2012, 22, 1033 RSC.
- M. J. Khan, S. Qayyum, F. Alam and Q. Husain, Nanotechnology, 2011, 22 DOI:10.1088/0957-4484/22/45/455708.
- S. Eigler, C. Dotzer and A. Hirsch, Carbon, 2012, 50, 3666 CrossRef CAS.
- T. N. Narayanan, Z. Liu, P. R. Lakshmy, W. Gao, Y. Nagoka, D. S. Kumar, L. R. Vajtai and P. M. Ajayan, Carbon, 2012, 50, 1338–1345, DOI:10.1016/j.carbon.2011.11.005.
- M. Mahdevi, M. B. Ahmad, M. J. Haron, F. Namvar, B. Nadi, M. Z. A. Rahman and J. Amin, Molecules, 2013, 18, 7533 CrossRef PubMed.
- S. A. Ansari and Q. Husain, J. Mol. Catal. B: Enzym., 2011, 70, 119 CrossRef CAS.
- J. F. Zhan, S. T. Jiang and L. J. Pan, Braz. J. Chem. Eng., 2013, 30, 721 CrossRef CAS.
- S. G. Valerio, J. S. Alves, M. P. Klien, R. C. Rodrigues and P. F. Hertz, Carbohydr. Polym., 2013, 92, 462 CrossRef CAS PubMed.
- P. Monsan, J. Mol. Catal., 1978, 3, 371 CrossRef CAS.
- C. Mateo, R. Monti, B. C. C. Pessela, M. Feuntes, R. Toress, J. M. Guisan and R. F. Lafuente, Biotechnol. Prog., 2004, 20, 1259 CrossRef CAS PubMed.
- A. Park and D. Oh, Appl. Microbiol. Biotechnol., 2010, 85, 1427 CrossRef CAS PubMed.
- P. Ye, J. Jiang and Z. K. Xu, Colloids Surf., B, 2007, 60, 62 CrossRef CAS PubMed.
- T. Haider and Q. Husain, Int. J. Biol. Macromol., 2007, 41, 72 CrossRef CAS PubMed.
- H. Bayraktar, M. Serilmiz, T. Karakas, E. B. Celem and S. Onal, Int. J. Biol. Macromol., 2011, 49, 855 CrossRef CAS PubMed.
- N. Singh and A. M. Kayastha, Carbohydr. Res., 2012, 358, 61 CrossRef CAS PubMed.
- K. M. Vnenchak and K. Rokosz, Folia Biol., 1997, 45, 153 Search PubMed.
- R. R. Tice, E. Agurell, D. Erson, B. Burlinson, A. Hartmann, H. Kobayashi, Y. Miyamae, E. Rojas, J. C. Rvu and Y. F. Sasaki, Environ. Mol. Mutagen., 2000, 35, 206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06960f |
| This journal is © The Royal Society of Chemistry 2016 |