
Diversity oriented synthesis of benzimidazole-based biheterocyclic molecules by combinatorial approach: a critical review
Abstract
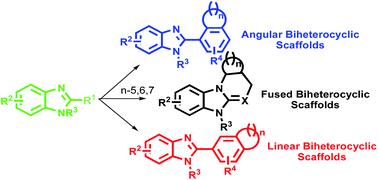
Heterocyclic compounds play a major role in drug discovery processes as these are the common structural motif for 80% of the marketed drugs by US retail sales in 2014. Benzimidazole and its derivatives are regarded as important heterocyclic motifs that exhibit a wide range of pharmaceutical applications including anticancers, antihypertensives, antivirals, antifungals and anti-HIVs. In view of their wide ranging bioactivities, it is imperative to focus on the synthesis of heterocyclic molecules containing benzimidazole as one of the moieties. This review focuses on the application of solid supports for the synthesis of different classes of benzimidazole-based biheterocyclic molecules in the last decade. In this review paper, detailed synthetic steps involved for the synthesis of benzimidazole-based biheterocyclic molecules, are shown in the schemes. The synthesis covers linear, angular and fused benzimidazole-based biheterocyclic molecules by a combinatorial approach. In addition, in the introduction section we shed some light on the use of biheterocyclic molecules as current drugs and open a new avenue for the utilization of these biheterocyclic molecules for pharmaceutical applications.
 Please wait while we load your content...
Please wait while we load your content...

