
Access to steroidal pyridazines via modified thiohydrazides†
Abstract
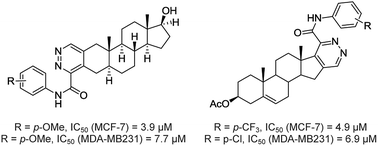
An approach to steroids annulated with pyridazines via cascade imination/electrocyclization of chlorovinyl aldehydes with oxamic acid thiohydrazides is disclosed. A mechanistic rationalization was performed using real-time 1H NMR spectroscopy and computational studies. A series of 18-nor-5α-androsta-2,13-diene[3,2-d]pyridazines, androsta-2-ene[3,2-d]pyridazines and Δ1,3,5(10)-estratrieno[16,17-d]pyridazines were synthesized from native hormones. These compounds were screened for cytotoxicity against the human estrogen-responsive breast cancer cell line MCF-7 and the estrogen-independent breast cancer cell line MDA-MB-231. The structure–activity relationship analysis revealed that the annulation of the pyridazine moiety to the A-ring of the 17β-hydroxy-5α-androsta-2-ene core provides high antiproliferative activity. Compounds 7a and 10b exhibited higher antiproliferative potency than the drug cisplatin. 5α-Androsta-2-ene[3,2-d]pyridazine 10c showed good selectivity against the MCF-7 breast cancer cells.
 Please wait while we load your content...
Please wait while we load your content...

