
Heteroatom-doped mesoporous carbons as efficient adsorbents for removal of dimethoate and omethoate from water
Abstract
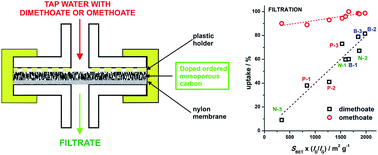
Extensive use of organophosphate pesticides (OPs) invokes development of efficient procedures for their removal from the environment. By introducing low levels (<1 at%) of B, N or P into the structure of mesoporous carbons, we have produced a series of materials with different surface chemical composition, textural properties and level of structural disorder. These adsorbents were applied for removal of dimethoate and its oxo-analogue omethoate from aqueous solutions under batch adsorption conditions and by filtration using modified nylon membrane filters. Adsorption capacities up to 164 mg g−1 were measured, with OPs uptake typically above 80% for dimethoate concentration as high as 5 × 10−3 mol dm−3. After the adsorption, neurotoxic effects of OP-containing water samples were significantly reduced or completely removed. The level of structural disorder was identified as a key parameter for efficient removal of dimethoate and omethoate while in the filtration experiments surface area of adsorbents also played an important role. While the presented research appeals to new fundamental studies of OP–carbon surface interactions, it also indicates a possible strategy in designing new efficient adsorbents for OPs removal from water.
 Please wait while we load your content...
Please wait while we load your content...

