
Electronically excited state of neutral/protonated, indole/5-hydroxyinodole–water clusters: a theoretical study†
 *a
*a
Abstract
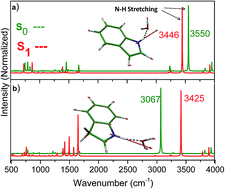
The second-order approximate coupled-cluster (RI-CC2) method was employed to investigate photoinduced hydrogen-bond weakening or strengthening in neutral and protonated indole–, 5-hydroxyindole–water clusters. In addition to the protonation effect on the electronic structure of 5-hydroxyindole, the intermolecular H-bond weakening or strengthening of selected systems in the S1 excited state has been investigated. According to our calculated results, it has been predicted that the electronic excitation effect on the hydrogen-bond strength in protonated clusters is essentially more pronounced than neutral analogues. Also, a charge transfer character of the excited state over the chromophore moiety can be suggested for interpreting the excited state dynamics of H-bonds in protonated complexes. Moreover, it has been predicted that protonation is accompanied by a strong red-shift effect (∼1.10 eV) on the S1–S0 transition energy of 5-hydroxyindole.
 Please wait while we load your content...
Please wait while we load your content...

