
Enhancing the antitumor effect of methotrexate in intro and in vivo by a novel targeted single-walled carbon nanohorn-based drug delivery system†
Ran Wanga,
Hongjing Cuia,
Junling Wanga,
Nannan Lia,
Qian Zhaoa,
Ying Zhoua,
Zhiyi Lva and
Wenying Zhong*ab
aDepartment of Analytical Chemistry, China Pharmaceutical University, Nanjing, China. E-mail: wyzhong@cpu.edu.cn
bDepartment of Medicinal Chemistry, China Pharmaceutical University, Nanjing 210009, PR China
First published on 6th May 2016
Abstract
The present research reports a smart multifunctional oxidized single-wall carbon nanohorns (oxSWNHs) drug delivery system (DDS) which could enhance the anti-tumor effect of methotrexate (MTX). In this DDS (MTX@oxSWNHs–PEG–Tf), oxSWNHs loaded MTX was wrapped with DSPE–PEG–NH2 to prolong blood circulation half-life and target tumors with the help of covalent grafting of transferrin (Tf). The obtained DDS was characterized by scanning electron microscopy, UV-vis spectrophotometry, size distribution analysis and Z-potential measurement. Cell uptake studies on MAD-MB-231 and HepG2 cell lines confirmed that the MTX@oxSWNHs–PEG–Tf exerted higher antitumor activity compared with free drug or non-targeted counterpart. The in vivo antitumor efficacy of MTX@oxSWNHs–PEG–Tf was much stronger than that of the free drug group by comparing the change of tumor volume and weight. Toxicity studies showed little perceivable cardiotoxicity, hepatotoxicity and pulmonary toxicity of MTX@oxSWNHs–PEG–Tf, indicating that this new tumor-targeting DDS significantly enhanced the anti-tumor effect of MTX and decreased its side effects.
1 Introduction
Methotrexate (MTX) is a folate antagonist which is widely used in therapies for human malignancies due to its role in DNA synthesis interruption. However, a series of its dose-related side effects limit the clinical use of the drug. Some studies found out that the limited use of MTX was mainly due to the short plasma half-life and low drug concentration in targeted tissues.1–3 Great effort was made to extend drug release and maintain adequate drug dose in tumors. For example, Nevozhay D. et al. developed a dextran conjugated with MTX, which prolonged the blood retention and had a higher accumulation in mouse model of leukemia;4 Karasulu HY et al. used microemulsion loaded MTX, which reached a high cytotoxicity on MCF-7, OVCAR and DUI145 cells;5 Lindgren et al. designed MTX conjugated with cell-penetrating peptides (YTA2 and YTA4), which was far more effective than free drug.6 In our study, we used single-walled carbon nanohorns (SWNHs) as drug carriers. The single-walled carbon nanohorn (SWNHs), which is an allotrope of graphite with a circular cone-like structure, is one of the most important members in nanomaterials.7 SWNHs, having a large surface area with a 2–5 nm in diameter and a 40–50 nm length, forms a spherical aggregate with a diameter of 80–100 nm. The big ball could get a ultrahigh drug loading capacity via π–π interaction8–11 and enhance the drug permeability and retention (EPR) effect, thus carrying sufficient drugs into tumor cells.12–15 Compared with other nanomaterials, SWNHs has a splendid advantage in that it could be prepared by laser ablation, prescinded from the influences of any metal impurities and without toxicity in vitro and in vivo.16,17 All the properties make SWNHs a superior drug carrier. However, the hydrophobicity of SWNHs has long been a major barrier in its biomedical application. To solve this problem, PEG was selected as a solubility enhancer to improve its hydrophilicity and prolong the half-life in serum.Chemical drugs have long been used in cancer treatments. However, their non-targeting attack to innocent proliferating cells leads to numerous side effects. Development of safe and effective target drug delivery system is in urgent need. In the past decade, a variety of materials such as folate, aptamers, antibodies and peptides have been investigated as target agents to accelerate drugs' target release and to reduce the side-effects.18–21 We have utilized some targeting ligands for tumor therapy in nanohorns drug delivery systems, such as VEGF and IGF-IR.22–24 Compared with these costly targeted agents, transferrin (Tf) not only has plenty of biological functions but also is used at much lower cost. Hence, we selected transferrin as the target agent to improve the drug concentration in tumor site and minimize the side effect of MTX.
Transferrin is a 78 kDa-monomeric glycoprotein, containing approximately 700 amino acids, 19 intrachain disulfide bonds and three carbohydrate side chains.25–27 The conformational changes of protein during iron binding have been demonstrated to prove a selective recognition made by the transferrin receptor (TfR). Transferrin's important functions are regulated by circulating iron. Overexpression of TfR on the surface of cancer cells goes along with the rapid cell proliferation, satisfying the high demand of Fe3+. The affinity for TfR in tumor sites is approximately 10–100 times higher than that in normal tissues. The high affinity makes transferrin a potential novel target for anticancer therapy.28–30 Compared with other single functional target agents, transferrin also has other effects such as erythropoiesis which promotes cell division, antimicrobial and prevents drug resistance.31–33 Wang and his coworkers showed the obvious reversal of drug resistance by conjugation of transferrin–doxorubicin through its saturation with gallium nitrate.34
In this study, we established a promising novel target drug delivery system based on oxSWNHs loaded anticancer drug-methotrexate, which was wrapped with solubility enhancer DSPE–PEG–NH2 (MW = 5000) and conjugated with the target ligand transferrin. We characterized the system in vitro and in vivo, which included the manner of drug release, the mechanism of target ligand influencing the tumor-targetability, as well as the drug efficacy.
2 Experimental
2.1 Materials and methods
The oxSWNHs (purity > 90%, lot no. K181-2) were purchased from NEC Corporation, Tokyo, Japan. MTX was purchased from Melone Pharmaceutical Co., Ltd. 1,2-Disteatoyl-sn-glycero-3-phosphoethanolamine–N-poly(ethylene glycol)-amine (DSPE–PEG, MW = 5000) was purchased from Laysan Bio, Inc, Alabama, American. Transferrin was purchased from YuanYe Company (Shanghai, China). Fluorescein isothiocyanate (FITC), 1-ethyl-(3-dimethyl-aminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and dialysis bags (MW = 3500) were purchased from Aladdin Reagent Corporation (Shanghai, China). Cell Counting Kit (CCK-8) was purchased from BeiDi Corporation (Nanjing, China). Dimethyl sulfoxide (DMSO) was purchased from Sinopharm Chemical Reagent Co. Dulbecco's Modified Eagle Medium cell culture medium was purchased from Gibco Co. Fetal bovine serum (FBS) was purchased from Giboco. The lung epithelial cancer cell line A549, breast cancer cell MAD-MB-231, H22 tumor cells and HUVECs cell were obtained from American Type Culture Collection (ATCC). The ICR mice were purchased from the Animal Care and Use Committee in China Pharmaceutical University. Enhanced BCA Protein Assay Kit was purchased from Beyotime Biotechnology Company.2.2 Synthesis of MTX@oxSWNHs and MTX@oxSWNHs–PEG
In a standard experiment, 0.5 mg oxSWNHs were dissolved in 1 mL anhydrous DMSO in an ampoule bottle by sonicating for 2 h. Next, 5 mg MTX was added to the oxSWNHs solution by sonicating for 2 h. Then the dispersive solution was left to magnetic stirring for 12 h followed by centrifugation and the supernatant was reserved. The obtained precipitation was washed with anhydrous DMSO and de-ionized water by centrifugation (four times at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min) to get rid of unbound MTX and subjected to freeze-drying. The unbound MTX concentration in filtrate was measured at 302 nm by the UV-1800 spectrophotometer (Shimadzu Corp, Japan). The complex was referred to as MTX@oxSWNHs.
000 rpm for 15 min) to get rid of unbound MTX and subjected to freeze-drying. The unbound MTX concentration in filtrate was measured at 302 nm by the UV-1800 spectrophotometer (Shimadzu Corp, Japan). The complex was referred to as MTX@oxSWNHs.
The MTX@oxSWNHs (0.5 mg oxSWNHs) was dispersed in 2 mL of de-ionized water and vortexed for seconds. Thereafter, DSPE–PEG (3 mg) was added and the mixture was stirred for 2 h in room temperature. The MTX@oxSWNHs–PEG was recovered by centrifugation (twice at 12000 rpm for 15 min), and washed by de-ionized water.
2.3 Synthesis of PEG–Tf and tagged with FITC
In order to develop tumor-targeting drug delivery system, we conjugated transferrin as a targeting ligand. PEG–Tf conjugation was obtained by mixing 8 mg of transferrin, 6 mg of DSPE–PEG and 100 μL of EDC (1 mg mL−1) in 2 mL of de-ionized water, and gently stirring for 12 h at 4 °C. Thereafter, the mixture was dialyzed in Phosphate Buffered Saline (PBS) to remove EDC and obtain PEG–Tf. FITC was used to label the drug delivery system. The PEG–Tf was dispersed in carbonate buffer solution (pH = 9), added 100 μL of FITC (1 mg mL−1), and gently stirred for 12 h at 4 °C in darkness. Then we obtained the PEG–Tf–FITC. Similarly, we synthesized MTX@oxSWNHs–PEG–Tf–FITC as described in Section 2.2.2.4 Characterization
:100 w/w), and KBr pellets for analysis were obtained under high pressure.2.5 In vitro cumulative release of MTX
The release behavior of MTX@oxSWNHs–PEG–Tf was carried in PBS at pH 7.4 and pH 5.5 to mimic the physiological and tumor pH, respectively. Briefly, the MTX@oxSWNHs–PEG–Tf (0.5 mg oxSWNHs) was carried in a dialysis bag (Millipore, MWCO = 3500), suspended in 50 mL of PBS, 0.1 M. The release assay was incubated at 37 ± 0.5 °C and gentle stirred (100 rpm min−1). Aliquots were taken at different time points and replaced by same volume of fresh buffer. Finally, the absorbance of MTX was recorded using UV spectroscopy at 302 nm. The release of the free drug was measured by following the same way. All of the experiment procedures were performed in triplicate.2.6 Cell image and cell viability measurements
The human lung adenocarcinoma (A549) cell line, breast adenocarcinoma (MAD-MB-231) cell line, non-tumor cell line human umbilical vein endothelial cells (HUVECs) and normal human liver cell (ML-7702) were grown in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 100 μg mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C, 5% CO2 in 95% air humidified atmosphere.To evaluate the targeting effect of drug delivery system (MTX@oxSWNHs–PEG–Tf), cultured cells were exposed to 50 μg mL−1 of MTX@oxSWNHs–PEG–Tf–FITC and MTX@oxSWNHs–PEG–FITC respectively and incubated for 4 h at 37 °C. Cells were then washed by pre-heated PBS at 37 °C for three times. Images were captured with a fluorescence microscopy (Olympus IX71, Japan).
The WST assay was performed to evaluate the cell viability. Cell lines were seeded in 96 well plates and incubated for 12 h. A549, MAD-MB-231 and ML-7702 cell lines were exposed to MTX@oxSWNHs–PEG–Tf, MTX@oxSWNHs–PEG, MTX and oxSWNHs–PEG–Tf at a concentration range of 0.001–100 μg mL−1. The drug concentrations were determined based on the total amount of MTX loaded on oxSWNHs which was to parallel the results. Cell lines were then incubated for 48 h and 10 μg of CCK-8 solutions were added and incubated for 1 h. Finally, cell viability was quantified by a BioRad microplate reader at 450 nm.
2.7 Western blot
HepG2 cell treated with MTX@oxSWNHs–PEG–Tf, MTX@oxSWNHs–PEG, MTX and oxSWNHs–PEG–Tf were extracted in lysis buffer, resolved by SDS-PAGE and transferred to nitrocellulose membranes. Proteins were detected using specific antibodies directed against PARP-1 (1:1000, Cell Signaling Technology, USA), Caspase-3 (1:1000, Abcam, USA) and P53 (1:1000, Millipore, USA). The protein expression levels were normalized with GAPDH (1:5000, Shengxing, China). After washing with TBST, the membrane was incubated with HRP-conjugated secondary antibody (1:10000, KeyGen, China) for 1 h. The signal was detected by enhanced chemiluminescence (ECL, Millipore).
2.8 In vivo antitumor activity
Experiments were performed on male ICR mice weighing 18–21 g (Yangzhou University Laboratory Animal Center, (No SCXK (Su) 2013-0011)). Experiments were carried out in strict accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals, and approved by the Scientific Committee of the China Pharmaceutical University. Animals were acclimated at 25 ± 0.5 °C under natural light/dark cycle in the prior two days. Then 2 mL of H22 tumor cells (1 × 107 cells per mouse) was injected subcutaneously into ICR mice. When the tumor size reached 100–300 mm3, mice were randomly divided into eight groups (six mice per group). For pharmacodynamics assessment, animals were injected with MTX@oxSWNHs–PEG–Tf (high: 5 mg kg−1, low: 1.5 mg kg−1), MTX@oxSWNHs–PEG; (high: 5 mg kg−1, low: 1.5 mg kg−1), MTX (high: 5 mg kg−1, low: 1.5 mg kg−1), oxSWNHs–PEG–Tf and normal saline (NS), respectively. Saline treated group served as control.Tumor size and body weight were measured by Vernier caliper every other day. The equation to calculate tumor size was as follows: tumor volume (V) = D × d2/2 (d and D denoted the shortest diameter and longest diameter of the tumor, respectively). The equation of relative tumor volume was RTV = V/V0 (V is the volume of the tumor after measurement and V0 was the initial volume of tumor). The tumor growth inhibition (TGI) evaluated the drug delivery system's antitumor efficacy with the equation: TGI = (V of normal saline group − V of treatment group)/V of normal saline group × 100% (where V was the average volume of tumor).
2.9 Tumor tissue section for microscopic observation
After 14 days of drug treatment, all of the mice were sacrificed by cervical vertebra dislocation. Individual organs (viz. liver, kidney, heart, lungs and tumor) of the test groups and control group were fixed in 10% formalin and embedded in paraffin. Paraffin sections were cut into 5 mm thickness, and then were stained with hematoxylin–eosin (H–E) according to standard method for histological. Morphological changes and cells apoptosis levels were observed with a microscope (Olympus DX45, Japan) and photographed using a camera (Olympus DP72, Japan).3 Results and discussion
3.1 Morphology of oxSWNHs and MTX@oxSWNHs–PEG–Tf
The morphology of oxSWNHs was evaluated by TEM. The representative image of oxSWNHs is shown in Fig. 1(a). oxSWNHs in water have a roughly spherical shape and form aggregates. In order to improve hydrophilicity, biocompatibility of oxSWNHs and prolong the blood circulation time, we selected amphiphilic polymer DSPE–PEG–NH2 (MW = 5000) to non-covalently modify oxSWNHs.35–37 In DSPE–PEG–NH2 polymer, DSPE was the hydrophobic end to graft on the oxSWNHs while the amino group was the hydrophilic end dissolved in water. The modification of oxSWNHs by DSPE–PEG was through π–π stacking, van der Waals and hydrophobic interactions.38 | ||
| Fig. 1 TEM images of (a) oxSWNHs, (b) MTX@oxSWNHs–PEG, (c) 1 photograph of MTX@oxSWNHs dispersed in water, PBS and cell culture for seven days (top), 2 photograph of MTX@oxSWNHs–PEG dispersed in water, PBS and cell culture for seven days (bottom). | ||
TEM image of MTX@oxSWNHs–PEG showed smooth edges of oxSWNHs due to wrapping of DSPE–PEG on oxSWNHs (Fig. 1(b)). Fig. 1(c) was the photograph of MTX@oxSWNHs and MTX@oxSWNHs–PEG dispersed in water, PBS and cell culture for seven days. Obviously, MTX@oxSWNHs–PEG displayed better dispersion and stability than oxSWNHs after seven days.
3.2 Size and zeta potential
DLS could evaluate the dispersion abilities of DSPE–PEG. As shown in Table 1, the size of MTX@oxSWNHs–PEG significantly decreased. The oxSWNHs presented an average hydrodynamic size and zeta potential of 194.5 ± 3.0 nm and −32.7 ± 0.6 mV, respectively. Followed by loaded MTX, the average of hydrodynamic size increased along with the value of zeta potential. Once MTX@oxSWNHs linked with PEG, the hydrodynamic size decreased to 175.13 ± 3.7 nm, while zeta potential successively increased to −11.9 ± 0.6 mV, which may be caused by the free amine group of PEG. The conjugation of Tf mildly increased the hydrodynamic size, and surface polarity shifted to −16.9 ± 0.4 mV, which was due to the acylation reaction between PEG and Tf. MTX@oxSWNHs–PEG–Tf's hydrodynamic size was 180.6 ± 1.9 nm, exceeding the threshold for renal clearance; therefore the blood circulation time was prolonged, and the EPR effect was enhanced.39 Meanwhile, the PDI values were all below 0.2, indicating that all the groups had a relative homogeneity in size.| oxSWNHs (nm) | MTX@oxSWNHs (nm) | MTX@oxSWNHs–PEG (nm) | MTX@oxSWNHs–PEG–Tf (nm) | |
|---|---|---|---|---|
| Size | 194.5 ± 3.0 | 206.4 ± 3.7 | 175.1 ± 3.7 | 180.6 ± 1.9 |
| PDI | 0.197 ± 0.049 | 0.196 ± 0.062 | 0.124 ± 0.019 | 0.154 ± 0.042 |
| Zeta | −32.7 ± 0.6 | −24.0 ± 0.5 | −11.9 ± 0.6 | −16.9 ± 0.4 |
3.3 Load behavior study
MTX loaded onto oxSWNHs was monitored by UV spectrophotometry.40,41 As shown in the UV spectrogram, free MTX exhibited three characteristic absorptions which peaks at 260, 303, 370 nm respectively (Fig. 2). The absorption peak of oxSWNHs at 264 nm displayed with a broad band. In the spectrum of MTX@oxSWNHs, peaks were observed at 260, 290 and 370 nm. Compared with free drug, the peak valley between 260 nm and 290 nm was unconspicuous. Notably, the change in the UV-vis spectrum confirmed that MTX was loaded onto oxSWNHs.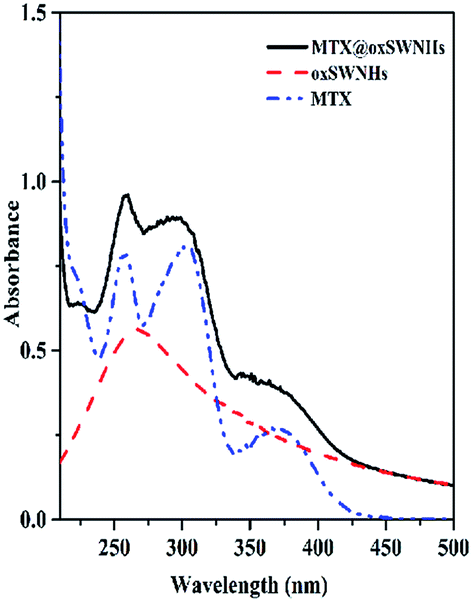 | ||
| Fig. 2 UV/vis absorbance spectra of MTX, oxSWNHs and MTX@oxSWNHs. | ||
The drug loading efficiency was determined by subtracting the amount of excessive free MTX in the supernatant after centrifugation. To quantify the concentration of unloaded-MTX, a calibration curve of MTX was drawn based on UV absorbance at 303 nm.42 As shown in Fig. 3, MTX's loading behavior was increased with the increase of the MTX dosage (from 1 mg to 20 mg). The loading proportion of MTX ranged from 20 to 151% (weight ratio of MTX/oxSWNHs), reaching the adsorption equilibrium. We chose a proper proportion of 1:10 where the amount of loaded MTX is 1.5 ± 0.023 mg mg−1 (MTX/oxSWNHs), and found out much more drugs loaded onto oxSWNHs than previous reports.24,43–45
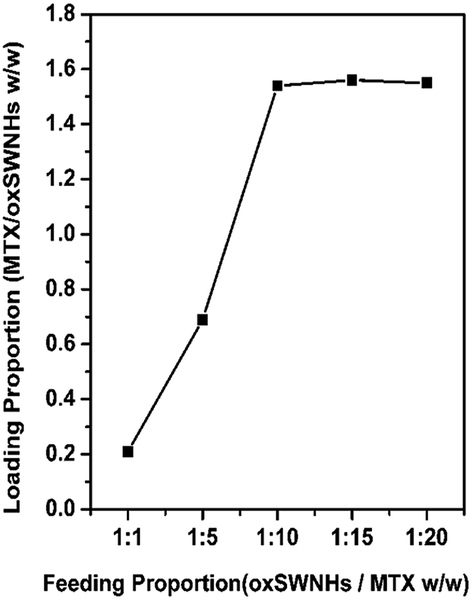 | ||
| Fig. 3 MTX loading at different feeding amounts of MTX (n = 3). | ||
The amount of MTX loaded onto oxSWNHs was also measured by the TGA. TGA measurement was carried out in temperature from 25 °C to 700 °C. TGA curves revealed that the oxSWNHs, MTX and MTX@oxSWNHs underwent about 10%, 69%, and 46% decreases in weight respectively. From the result of TGA, the weight of MTX loaded onto oxSWNHs was calculated to be 1.5 mg MTX per mg oxSWNHs, which is consistent with the results of UV-vis spectrum. The weight loss of MTX@oxSWNHs–PEG was 56.5%, while DSPE–PEG degraded completely. Therefore, the weight percent of DSPE–PEG was 54% of per mg oxSWNHs.
3.4 Target study and cell image
Tf's covalent bonding to PEG realized the target function of MTX@oxSWNHs–PEG–Tf. Previous studies reported that a PEG-like spacer insertion between nano-carrier and targeting ligands could improve the drug delivery system's hydrophilicity, thus increasing its accessibility towards receptor.46,47 In order to demonstrate the covalent linkage between Tf and DSPE–PEG, we operated FTIR measurements. Fig. 4 shows the spectra of Tf, PEG and PEG–Tf. PEG presented typical peaks at 3437 cm−1, 1649 cm−1 and 1110 cm−1, ascribable to the amine group, amide I and two alkyl ether (–CH2CH2O–) respectively. Transferrin conjugation is evidenced by the appearance in PEG–Tf spectrum. Compared with Tf, the typical peaks of PEG–Tf were from 1650 cm−1 to 1645 cm−1 attributed to amide I and 1529 cm−1 to 1556 cm−1 attributed to amide II. This shift was probably caused by their conformational changes after conjugation interaction.48,49 Compared with PEG, the peak at 1108 cm−1 corresponds to the presence of an –CH2CH2O– group. These results suggested that the conjugation of PEG–Tf was synthesized successfully. The XPS results (Fig. S1 and S2†) also confirmed the accomplishment of the drug delivery system.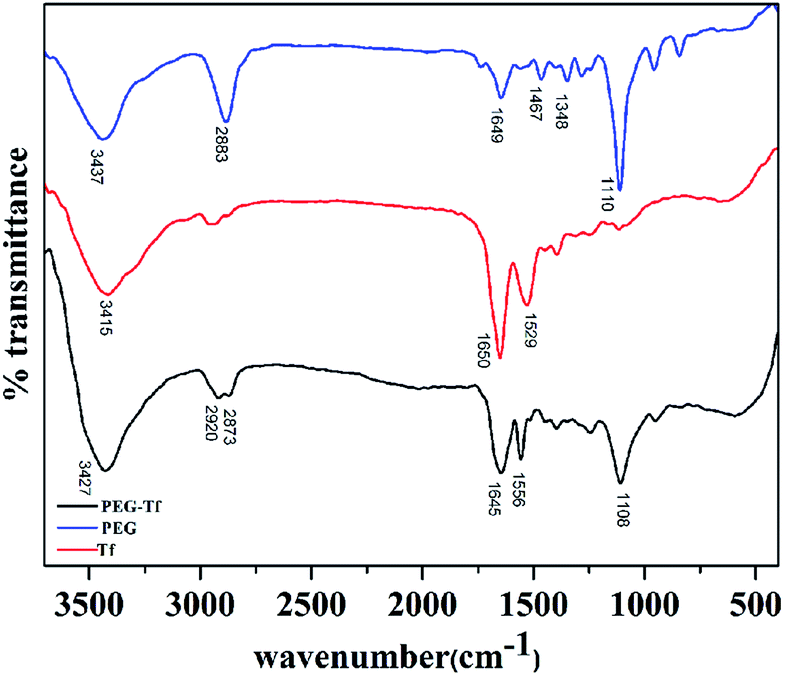 | ||
| Fig. 4 The FTIR absorbance spectra of Tf, PEG and PEG–Tf. | ||
BSA assays were performed on the supernatant of MTX@oxSWNHs–PEG–Tf to control the protein levels, quantifying 360 μg of Tf for 1 mg of MTX@oxSWNHs–PEG–Tf. Thus the ratio of Tf:PEG was about 1:1.5 (w/w).
The function of DDS on tumor targeting was examined by inverted fluorescence microscopy. FITC was used to label DDS and track the intracellular trafficking with green fluorescence. MTX@oxSWNHs–PEG–Tf–FITC was incubated with MAD-MB-231 tumor cell, HepG2 tumor cell and HUVEC cell for 4 h at 37 °C. In bright field (Fig. 5), the black particles were attached to tumor cells. A bright punctuation of green fluorescence in the dark field was also caught at the same location. However, there are few black particles attached to HUVE cell, and little fluorescence could be observed in the dark field as shown in the Fig. 5(c). The results clearly demonstrated that DDS selectively targeted to tumor cell MAD-MB-231 and HepG2 instead of HUVE cell. This may be due to the cellular uptake enhancement induced by the transferrin–transferrin receptor interaction.50 The transferrin receptor was overexpressed on the surface of tumor cell, but little in normal cells, thus providing that the Tf conjugated DDS could target to tumor tissues and selectively kill cancer cells.
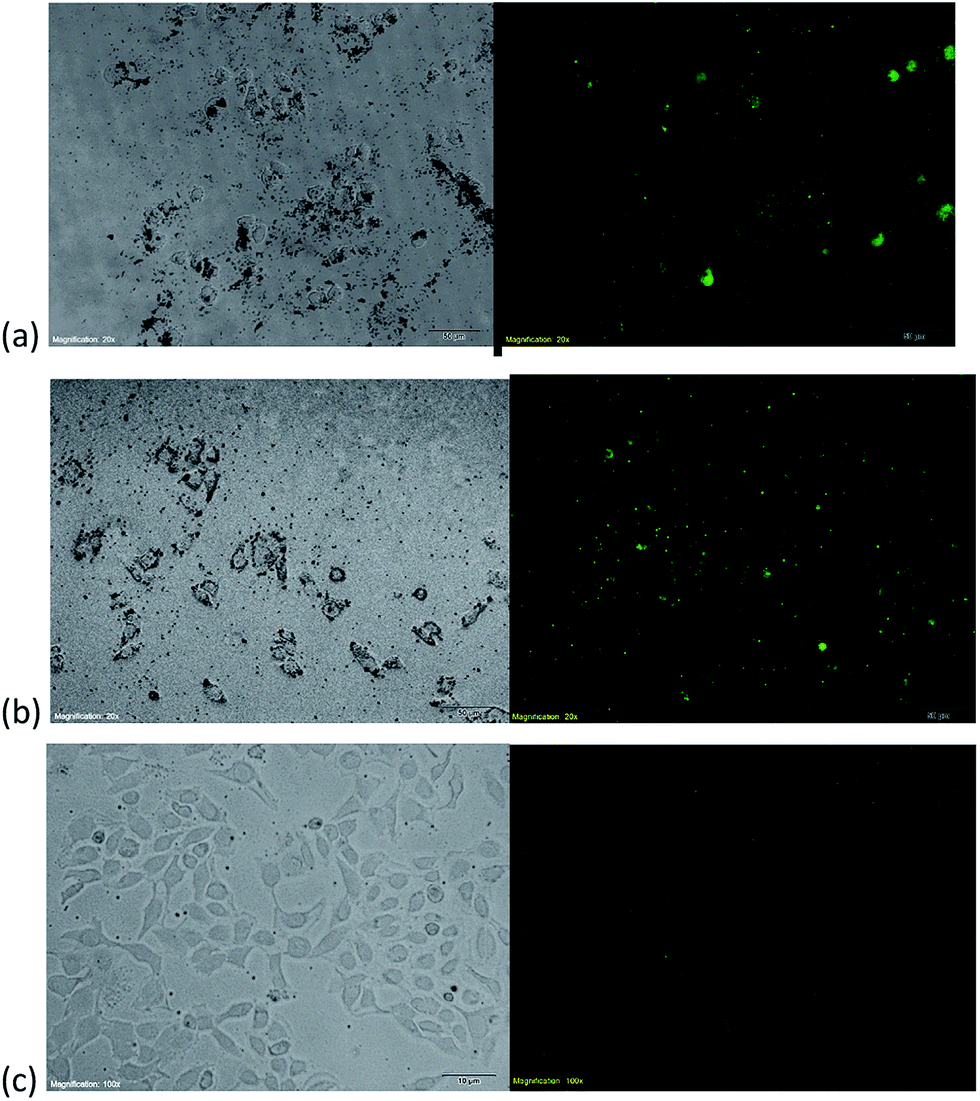 | ||
| Fig. 5 (a) Bright-field-microscopy image (left) and fluorescence microscopic images of MTX@oxSWNHs–PEG–Tf/FITC (right) in MAD-MB-231 cells. (b) Bright-field-microscopy image (left) and fluorescence microscopic images of MTX@oxSWNHs–PEG–Tf/FITC (right) in HepG2 cells. (c) Bright-field-microscopy image (left) and fluorescence microscopic images of MTX@oxSWNHs–PEG–Tf/FITC (right) in HUVE cells. | ||
3.5 Release behavior of MTX from oxSWNHs
The MTX release behavior from drug delivery system (DDS) was determined in phosphate buffer of pH 7.4 and pH 5.5, which was to mimic the normal physiological state and tumor microenvironment (Fig. 6) respectively. Free MTX was used as the control under the same condition. No matter in pH 5.5 or in pH 7.4, the free MTX underwent a rapid release in few hours, but the DDS's drug release rates were much slower than the free MTX, indicating that the DDS could transport more drug to the tumor sites than free MTX. The release profile of MTX from the DDS experienced two stages: the initial burst release and the latter slow release. The initial burst release of MTX from the DDS was probably due to the loosely attached MTX molecules, e.g., the MTX absorbed on other MTX which interacted with oxSWNHs.22,41 The latter slow phase of drug release was due to the detachment of MTX molecules from oxSWNHs. The DDS's drug release rate in pH 5.5 phosphate buffer (55.66% ± 3.15%) was somewhat higher than that in pH 7.4 phosphate buffer (49.01% ± 1.93%). The release profile revealed that drug in DDS would be released more rapidly in acid environment such as tumor sites than in the normal tissues. It is worthwhile to be noted that a pH value of 5.5 could ionize about 50% of the carboxylic groups of MTX, increasing the hydrophilicity and the electrostatic repulsion between the drug; thereby, it contributed to the drug release.51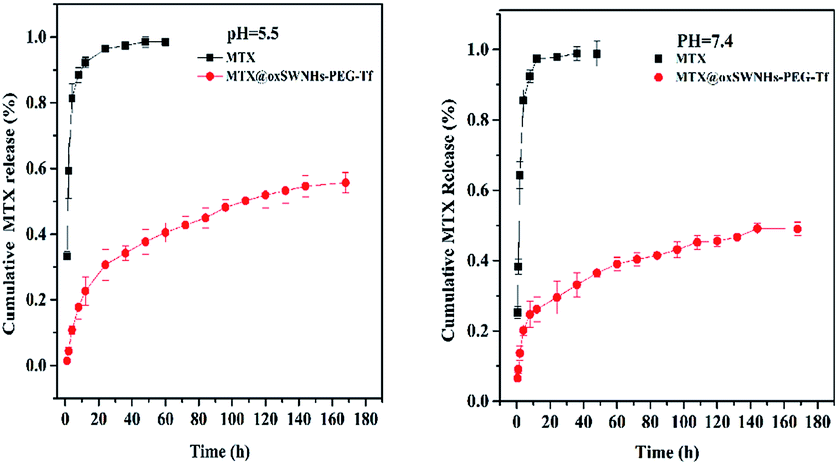 | ||
| Fig. 6 In vitro release profiles of MTX from different formulations (n = 3) (left: pH 5.5; right pH 7.4). | ||
3.6 Cytotoxicity and apoptosis study
The WST assays were performed to evaluate the cytotoxicity by measuring the metabolic activity of viable cells. The cytotoxicity of MTX@oxSWNHs–PEG–Tf, MTX@oxSWNHs–PEG and MTX were tested on tumor cells (MAD-MB-231 and HepG2) and normal liver cells (HL-7702). MTX deprived oxSWNHs–PEG–Tf was taken as control, and the concentrations of oxSWNHs–PEG–Tf were matched to the drug-loaded formulations (MTX@oxSWNHs–PEG–Tf, MTX@oxSWNHs–PEG). The results clearly revealed that all MTX formulations tested (MTX@oxSWNHs–PEG–Tf, MTX@oxSWNHs–PEG and MTX) exhibited dose-dependent reduction in cellular viability. As evidences displayed in Fig. 7, the oxSWNHs–PEG–Tf exhibited negligible cytotoxicity in cellular viability assay, even at the highest concentration. MTX@oxSWNHs–PEG–Tf and MTX@oxSWNHs–PEG exerted higher cytotoxicity than free drug in both cell lines. At the same time, the MTX@oxSWNHs–PEG–Tf exhibited higher antitumor activity than MTX@oxSWNHs–PEG. Table 2 showed the IC50 values of MTX@oxSWNHs–PEG–Tf were 1.27 in HepG2 cells, 1.80 in MAD-MB-231 cells and 34.35 in ML-7702 cells. Remarkably, DDS had the highest inhibition efficiency on tumor cells and minimum cytotoxicity on normal cells. The increased antitumor activities of drug-loaded formulations may be attributed to two points: (i) oxSWNHs drug delivery system could carry more drugs into cancer cells; (ii) increased affinity activity of transferrin–transferrin receptor to targeted drug delivery. The results on normal cell line revealed that MTX@oxSWNHs–PEG–Tf exerted lower cytotoxicity than MTX@oxSWNHs–PEG and MTX. That is likely due to fewer transferrin–transferrin receptors on the normal cell surface. At the same time, MTX exhibited higher toxicity than MTX@oxSWNHs–PEG, which may attribute to the sluggish release of MTX (from MTX@oxSWNHs–PEG) under neutral pH environments. | ||
| Fig. 7 (a) Cell inhibition of MAD-MB-231 cells after 48 h incubation. (b) Cell inhibition of HepG2 cells after 48 h incubation. (c) Cell inhibition of HL-7702 cells after 48 h incubation. | ||
| Sample examined | MTX@oxSWNHs–PEG–Tf | MTX@oxSWNHs–PEG | MTX |
|---|---|---|---|
| IC50 (μg mL−1), HepG2 | 1.27 | 2.96 | 6.71 |
| IC50 (μg mL), MAD-MB-231 | 1.80 | 3.80 | 7.05 |
| IC50 (μg mL−1), HL-7702 | 34.35 | 7.29 | 4.94 |
Western blot assays were performed to explore the mechanism of cell apoptosis inducing effect of MTX@oxSWNHs–PEG–Tf. Apoptotic proteins such as PARP-1, caspase-3 and P53 were detected in HepG2 cells, which were treated with oxSWNHs–PEG–Tf (A), MTX (B), MTX@oxSWNHs–PEG (C) and MTX@oxSWNHs–PEG–Tf (D). A gradient increase was detected among the groups, similar as results in cytotoxicity assays (Fig. 8).
 | ||
| Fig. 8 Induction of PARP-1, caspase-3 and P53 by oxSWNHs–PEG–Tf (A), MTX (B), MTX@oxSWNHs–PEG (C) and MTX@oxSWNHs–PEG–Tf (D) all treatment were about 1 μg mL−1 of MTX, in HepG2 cells. | ||
3.7 In vivo antitumor performance
The in vivo antitumor performance of the DDS was further evaluated with the ICR male mice models bearing subcutaneously inoculated H-22 tumor. Mices were treated with all kinds of clinical MTX injection with two doses normalized to 1.5 or 5 mg of MTX equivalent per kilogram of body weight, respectively. MTX@oxSWNHs–PEG–Tf, MTX@oxSWNHs–PEG, MTX and oxSWNHs–PEG–Tf were intratumorally injected into tumor-bearing mice every 3 days, and saline-treated groups were used as control. All of the mice were observed for clinical symptoms, and a caliper was used to measure tumor size every other day. Fig. 9 presented the tumor-growth profile of mice treated with different drug systems. After 14 days of treatment, the oxSWNHs–PEG–Tf group and saline-treated group both displayed a rapid and time-dependent growth, indicating the two treatments have parallel efficacy on tumor growth suppression. Compared with the control group, the oxSWNHs–PEG–Tf group displayed a slight antitumor activity. It is obvious in Fig. 9 that all groups exhibited dose-dependent changes of average relative tumor volumes (V/V0). In dose of 5 mg kg−1, the value of RTV was 9.08 ± 0.81 for the MTX group and 5.68 ± 0.87 for the MTX@oxSWNHs–PEG group, approximately 1.87 and 2.98 times less than those of the saline group. It is notable that the MTX@oxSWNHs–PEG inhibited tumor growth much more efficiently than the free MTX at the same dose. The results might be caused by the induction of PEG chain which prolonged the drug blood circulation and delivered drugs continuously to the tumor location. Among all the groups, the group treated with MTX@oxSWNHs–PEG–Tf showed significant higher antitumor efficiency, with an RTV of 4.61 ± 0.54 (5 mg kg−1), approximately 3.68 times less than the control's. The high antitumor efficacy of MTX@oxSWNHs–PEG–Tf may be attributed to its tumor-specific accumulation. Tf enhanced the activity of transferrin–transferrin receptor to targetedly deliver drug to the tumor, hence achieving fantastic antitumor effect. After drug treatment, tumors were excised from sacrificed mice. The excised sarcomas weight served as the intuitive evidence of antitumor effect of each group showed in Fig. 10. During the entire course of study, all the groups showed a growth trend in mice's body weight. Mice in the free drug group (5 mg kg−1) exhibited a weak, sluggish performance and congregated at the corner of the cage; this situation was not observed in any other group. This phenomenon should be explained by that the DDS had lower side effects than free drug. In a word, the targeted DDS exhibited a highly effective antitumor performance and little side effects. | ||
| Fig. 9 (a) Pharmacodynamic assessment in rats of tumor growth inhibition (V/V0) (n = 8). (b) Pharmacodynamic assessment in rats of mean body weight of mice (n = 8). | ||
 | ||
| Fig. 10 Photograph of tumor taken from different group (right) and the average weight of tumor (left). | ||
3.8 Histopathological examination
In order to evaluate the necrosis levels of different organs of test groups (MTX@oxSWNHs–PEG–Tf 5 mg kg−1, MTX 5 mg kg−1, oxSWNHs–PEG–Tf and normal saline), mice were sacrificed after 14 days of treatments. Organ slices were stained with hematoxylin and eosin (HE). All the groups did not reveal obvious cardiac, lung or hepatic damages except the MTX group, which promoted moderate liver inflammatory cells infiltration and extramedullary hematopoiesis. This result might be the side effect of drug. In addition, the injected MTX@oxSWNHs–PEG–Tf did not cause any significant lesion to the tested organs. As shown in Fig. 11, tumor histological images showed a highest necrotic rate in the group of MTX@oxSWNHs–PEG–Tf. The necrotic rate in MTX group is obviously higher than that in the control and deprived MTX group. These results confirmed the superior treatment efficiency of MTX@oxSWNHs–PEG–Tf. | ||
| Fig. 11 Representative H & E stained images of major organs including heart, liver, spleen, lung, and kidney and tumor resected from mice on the 14th day after treatments with saline, oxSWNHs–PEG, MTX, MTX@oxSWNHs–PEG (5 mg kg−1), MTX@oxSWNHs–PEG–Tf (5 mg kg−1). | ||
4 Conclusions
In conclusion, our study presented a novel multifunctional drug delivery system based on oxSWNHs. The novel system (MTX@oxSWNHs–PEG–Tf) was blended with three functional moieties: the chemotherapeutic agent MTX, the modifier PEG and the targeting agent Tf. The drug delivery system of MTX@oxSWNHs–PEG–Tf was characterized by UV, IR, TGA, TEM, DLS and BCA. The MTX@oxSWNHs–PEG–Tf was successfully loaded with high dose of MTX (1.50 ± 0.023 mg mg−1), triggering intracellular drug accumulative releases. The system was coated with PEG to ameliorate water solubility and biocompatibility, which provided the carrier system with particle size of 180.6 ± 1.9 nm and zeta potential of −16.9 ± 0.4 mV. The in vitro release behavior experiment exhibited that DDS had a distinctly more sustainable release behavior than free drugs. DDS also had a higher release at pH of 5.5 than at pH of 7.4. In vitro and in vivo studies proved that due to the high tumor avidity, DDS could accurately target tumor site, thus enhancing its antitumor efficacy, and alleviating its deleterious effects against normal tissues at the same time. Overall, the MTX@oxSWNHs–PEG–Tf is a promising DDS in clinical cancer therapies which could be of widespread applications in the field of cancer.Acknowledgements
This work was supported with financial support from the National Natural Science Foundation of China (No. 81173023), and supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- D. Banerjee, P. Mayer-Kuckuk, G. Capiaux, T. Budak-Alpdogan, R. Gorlick and J. R. Bertino, Biochim. Biophys. Acta, Mol. Basis Dis., 2002, 1587, 164–173 CrossRef CAS.
- C. Fiehn, Clin. Exp. Rheumatol., 2009, 28, S40–S45 Search PubMed.
- M. P. Iqbal, J. Pak. Med. Assoc., 1998, 48, 341–342 CAS.
- D. Nevozhay, R. Budzynska, M. Jagiello, U. Kanska, M. S. Omar, A. Opolski, J. Wietrzyk and J. Boratynski, Anticancer Res., 2006, 26, 2179–2186 CAS.
- H. Karasulu, G. Kantarcı, B. Karaca, V. Armagan, T. Güneri and E. Göker, Drug Dev. Res., 2009, 70, 49–56 CrossRef CAS.
- M. Lindgren, K. Rosenthal-Aizman, K. Saar, E. Eiríksdóttir, Y. Jiang, M. Sassian, P. Östlund, M. Hällbrink and Ü. Langel, Biochem. Pharmacol., 2006, 71, 416–425 CrossRef CAS PubMed.
- M. Horie, L. K. Komaba, H. Fukui, H. Kato, S. Endoh, A. Nakamura, A. Miyauchi, J. Maru, E. Miyako and K. Fujita, Carbon, 2013, 54, 155–167 CrossRef CAS.
- M. Bottini, F. Cerignoli, M. I. Dawson, A. Magrini, N. Rosato and T. Mustelin, Biomacromolecules, 2006, 7, 2259–2263 CrossRef CAS PubMed.
- K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, S. Wieckowski, J. Luangsivilay, S. Godefroy, D. Pantarotto, J.-P. Briand and S. Muller, Nat. Nanotechnol., 2007, 2, 108–113 CrossRef CAS PubMed.
- Z. Liu, W. Cai, L. He, N. Nakayama, K. Chen, X. Sun, X. Chen and H. Dai, Nat. Nanotechnol., 2007, 2, 47–52 CrossRef CAS PubMed.
- G. Pastorin, Pharm. Res., 2009, 26, 746–769 CrossRef CAS PubMed.
- S. Iijima, M. Yudasaka, R. Yamada, S. Bandow, K. Suenaga, F. Kokai and K. Takahashi, Chem. Phys. Lett., 1999, 309, 165–170 CrossRef CAS.
- N. Li, Z. Wang, K. Zhao, Z. Shi, Z. Gu and S. Xu, Carbon, 2010, 48, 1580–1585 CrossRef CAS.
- L. Mercatelli, E. Sani, G. Zaccanti, F. Martelli, P. Di Ninni, S. Barison, C. Pagura, F. Agresti and D. Jafrancesco, Nanoscale Res. Lett., 2011, 6, 1–9 CrossRef PubMed.
- S. Zhu and G. Xu, Nanoscale, 2010, 2, 2538–2549 RSC.
- D. Chen, C. Wang, F. Jiang, Z. Liu, C. Shu and L.-J. Wan, J. Mater. Chem. B, 2014, 2, 4726–4732 RSC.
- J. Miyawaki, M. Yudasaka, T. Azami, Y. Kubo and S. Iijima, ACS Nano, 2008, 2, 213–226 CrossRef CAS PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS PubMed.
- F. X. Gu, R. Karnik, A. Z. Wang, F. Alexis, E. Levy-Nissenbaum, S. Hong, R. S. Langer and O. C. Farokhzad, Nano Today, 2007, 2, 14–21 CrossRef.
- L. M. Weiner, R. Surana and S. Wang, Nat. Rev. Immunol., 2010, 10, 317–327 CrossRef CAS PubMed.
- N. Li, Q. Zhao, C. Shu, X. Ma, R. Li, H. Shen and W. Zhong, Int. J. Pharm., 2015, 478, 644–654 CrossRef CAS PubMed.
- X. Ma, C. Shu, J. Guo, L. Pang, L. Su, D. Fu and W. Zhong, J. Nanopart. Res., 2014, 16, 1–14 CAS.
- Q. Zhao, N. Li, C. Shu, R. Li, X. Ma, X. Li, R. Wang and W. Zhong, J. Nanopart. Res., 2015, 17, 1–14 CrossRef.
- P. T. Gomme, K. B. McCann and J. Bertolini, Drug Discovery Today, 2005, 10, 267–273 CrossRef CAS PubMed.
- W. Guo, W. Zheng, Q. Luo, X. Li, Y. Zhao, S. Xiong and F. Wang, Inorg. Chem., 2013, 52, 5328–5338 CrossRef CAS PubMed.
- D. H. Jiang, Y. Ke, Y. Z. Cheng, K. P. Ho and Z. M. Qian, Dev. Neurosci., 2002, 24, 94–98 CrossRef CAS PubMed.
- P. Dowling, V. Palmerini, M. Henry, P. Meleady, V. Lynch, J. Ballot, G. Gullo, J. Crown, M. Moriarty and M. Clynes, BBA Clinical, 2014, 2, 24–30 CrossRef PubMed.
- R. Koka and O. B. Ioffe, Semin. Diagn. Pathol., 2013, 30, 321–328 CrossRef PubMed.
- D. R. Richardson and P. Ponka, Biochim. Biophys. Acta, Rev. Biomembr., 1997, 1331, 1–40 CrossRef CAS.
- M. T. Andrés and J. F. Fierro, Antimicrob. Agents Chemother., 2010, 54, 4335–4342 CrossRef PubMed.
- L. J. Jensen, M. Kuhn, M. Stark, S. Chaffron, C. Creevey, J. Muller, T. Doerks, P. Julien, A. Roth and M. Simonovic, Nucleic Acids Res., 2009, 37, D412–D416 CrossRef CAS PubMed.
- M. Szwed, A. Matusiak, A. Laroche-Clary, J. Robert, I. Marszalek and Z. Jozwiak, Toxicol. In Vitro, 2014, 28, 187–197 CrossRef CAS PubMed.
- W. Faulk, Methods and materials for targeting and affecting selected cells, UtilityPatent, US2004220086A1, 2004 Search PubMed.
- N. Rubio, M. A. Herrero, M. Meneghetti, Á. Díaz-Ortiz, M. Schiavon, M. Prato and E. Vázquez, J. Mater. Chem., 2009, 19, 4407–4413 RSC.
- C. Shu, J. Zhang, J. Ge, J. H. Sim, B. G. Burke, K. A. Williams, N. M. Rylander, T. Campbell, A. Puretzky and C. Rouleau, Chem. Mater., 2009, 22, 347–351 CrossRef.
- M. Yang, M. Wada, M. Zhang, K. Kostarelos, R. Yuge, S. Iijima, M. Masuda and M. Yudasaka, Acta Biomater., 2013, 9, 4744–4753 CrossRef CAS PubMed.
- H. Dai and R. J. Chen, Noncovalent sidewall functionalization of carbon nanotubes, Utilitypatent, US8029734, 2011 Search PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- M. Das, S. R. Datir, R. P. Singh and S. Jain, Mol. Pharmaceutics, 2013, 10, 2543–2557 CrossRef CAS PubMed.
- D. R. Nogueira, L. Tavano, M. Mitjans, L. Pérez, M. R. Infante and M. P. Vinardell, Biomaterials, 2013, 34, 2758–2772 CrossRef CAS PubMed.
- N. T. T. Tran, T.-H. Wang, C.-Y. Lin and Y. Tai, Biochem. Eng. J., 2013, 78, 175–180 CrossRef CAS.
- J. Hrkach, D. Von Hoff, M. M. Ali, E. Andrianova, J. Auer, T. Campbell, D. De Witt, M. Figa, M. Figueiredo and A. Horhota, Sci. Transl. Med., 2012, 4, 128ra139 Search PubMed.
- Y. Mi, Y. Liu and S.-S. Feng, Biomaterials, 2011, 32, 4058–4066 CrossRef CAS PubMed.
- T. Murakami, J. Fan, M. Yudasaka, S. Iijima and K. Shiba, Mol. Pharmaceutics, 2006, 3, 407–414 CrossRef CAS PubMed.
- M. Das, D. Mishra, P. Dhak, S. Gupta, T. K. Maiti, A. Basak and P. Pramanik, Small, 2009, 5, 2883–2893 CrossRef CAS PubMed.
- M. Das, D. Mishra, T. Maiti, A. Basak and P. Pramanik, Nanotechnology, 2008, 19, 415101 CrossRef PubMed.
- J.-H. Park, J. Park, U. Dembereldorj, K. Cho, K. Lee, S. I. Yang, S. Y. Lee and S.-W. Joo, Anal. Bioanal. Chem., 2011, 401, 1631–1639 CrossRef PubMed.
- U. Dembereldorj, E.-O. Ganbold, J.-H. Seo, S. Y. Lee, S. I. Yang and S.-W. Joo, Vib. Spectrosc., 2012, 59, 23–28 CrossRef CAS.
- G. Ciofani, S. Del Turco, G. G. Genchi, D. D'Alessandro, G. Basta and V. Mattoli, Int. J. Pharm., 2012, 436, 444–453 CrossRef CAS PubMed.
- P. Calvo, C. Remuñan-López, J. L. Vila-Jato and M. J. Alonso, Pharm. Res., 1997, 14, 1431–1436 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06667d |
| This journal is © The Royal Society of Chemistry 2016 |