
Astridia velutina-like S, N-codoped hierarchical porous carbon from silk cocoon for superior oxygen reduction reaction†
Yixuan Wanga,
Yongpeng Lei*abc and
Huaping Wanga
aKey Laboratory of High Performance Fibers & Products, Ministry of Education, Donghua University, Shanghai, P. R. China 201620. E-mail: lypkd@163.com; leiyongpeng@nudt.edu.cn; Fax: +86 731 84575118; Tel: +86 731 84575118
bCollege of Basic Education, National University of Defense Technology, Changsha, Hunan 410073, P. R. China
cKey Laboratory of Lightweight and Reliability Technology for Engineering Vehicle, College of Hunan Province, Changsha, Hunan 410114, P. R. China
First published on 29th July 2016
Abstract
The development of heteroatom-doped nanostructured metal-free carbon materials for the oxygen reduction reaction (ORR) is a critical challenge. In this work, through a one-step heat treatment, Astridia velutina-like S, N-codoped micro–mesoporous carbon (SNC-x) was synthesized from silk cocoon. The product was fully characterized by physicochemical and electrochemical techniques. The sample obtained at 800 °C possessed a unique hierarchical porous structure with 9.75 at% N and 1.79 at% S content, providing abundant active sites and thus facilitating efficient mass transport. The ORR activity was observed with an onset potential of 0.853 V (vs. RHE), comparable current density (4.5 mA cm−2 at 0 V) to commercial Pt/C as well as good methanol tolerance and electrochemical stability in alkaline electrolyte. Benefiting from the cheap biomass precursor and unique structural features, the product may serve as a potential candidate for energy storage, conservation and environmental applications, etc.
Introduction
The proton-exchange membrane fuel cell (PEMFC) is among the most efficient energy conversion devices for energy applications such as automotive, communication, power stations and so on. The oxygen reduction reaction (ORR) at the cathode has aroused considerable attention due to its great significance.1,2 The commercial Pt/C catalyst has been widely used due to its comparatively low overpotential and high current density. However, apart from the limited natural reserves and high cost of Pt, the sluggish kinetics, poor stability and anode crossover in electrochemical environments have severely encumbered the extensive application of Pt/C.3–5 Thus, low cost electrocatalysts with comparable or even better ORR activity and durability are highly desired.6As known, carbon materials have been widely studied as catalyst and support for electrochemical application due to large surface area, excellent conductivity and chemical stability, etc.7,8 Intensive research efforts are concentrated on the preparation of carbon based metal-free ORR catalysts, such as graphene,9–13 carbon nanotubes,14 nanoporous carbon,15,16 carbon fibers17–19 and carbon nanosheets,19,20 etc. Among them, doped graphene has been widely employed as both catalyst supports and metal-free catalysts in ORR. As far as we know, heteroatom doping (including N, B, P, S and I) break the electroneutrality of the adjacent carbon atoms and introduce plenty of defects, which is favorable for oxygen adsorption and thus improve the ORR activity.21 What's more, compared to single-doped carbon, the dual-even tri-doped carbon exhibited much higher activity and durability due to the synergetic effect between different elements.22,23
Notably, sustainable biomasses have been extensively studied as precursors for the preparation of N-doped carbons,24–26 illustrating special advantages such as extensive availability, environmental friendliness, easy preparation and low cost. In this vein, highly porous, conductive, heteroatom doped-carbons with high surface area were obtained by pyrolyzing human hair.27 The nitrogen-doped graphene (NG) was synthesized via pyrolysis of crystal sugar in the presence of urea as metal-free ORR electrocatalyst.28 Lately, Wu and co-workers29 adopted biopolymer silk cocoon as precursor to fabricate binder-free supercapacitor electrode. The hierarchical porous silk cocoon-derived carbon with interconnected network structure possessed large pore volume and high specific surface area, thus facilitating the ionic transportation.29
Herein, we demonstrate a facile but effective pathway to synthesize S, N-codoped micro–mesoporous carbon from silk cocoon. The one-step codoping of S and N element was realized by simply controlling the thermolysis temperature in the presence of thiourea. It is worth mentioning that KOH was employed to create 3D hierarchical porous structure. The product shows good ORR catalytic behaviour with onset potential of 0.853 V (vs. RHE), comparable current density, high methanol tolerance to Pt/C in alkaline media. The superior ORR activity can be mainly attributed to the unique pore structure and optimized dual-doping. This kind of 3D interconnected networks has tremendous potential in supercapacitor,30 lithium ion battery,31 and so on.
Experimental
Materials
Silk cocoon was supplied by a local market in Changsha, P. R. China and directly used as the starting material without any pre-treatment. Potassium hydroxide and ethanol were bought from Changsha Huihong Co. Ltd., China. Hydrochloric acid was purchased from Beijing Chemical Works., China. Thiourea was purchased from Sino-pharm Chemical Reagent Co. Ltd., China. All reagents were used as received. Deionized water used throughout the experiments was prepared by a Millipore-Q water system. High purity nitrogen was purchased from Changsha Xianggang Co. Ltd., China.Preparation of SNC-x
The S, N co-doped carbon materials were prepared by the following procedures: firstly, 0.1 g silk cocoon, 0.8 g thiourea and 0.3 g KOH were mixed and grounded in an agate mortar. The mixture was then transferred into a tube furnace and heated to the target temperature with a holding time of 2 h (3 °C min−1) under N2 atmosphere. After naturally cooling down to room temperature, the sample was washed in hydrochloric acid until pH = 7 and lastly dried at 80 °C in an oven overnight. The samples heat-treated under different temperatures were designated as SNC-x, where x means the corresponding temperature (e.g. SNC-800 means the samples heat treated at 800 °C). The preparation of SNC-x is displayed in Fig. 1.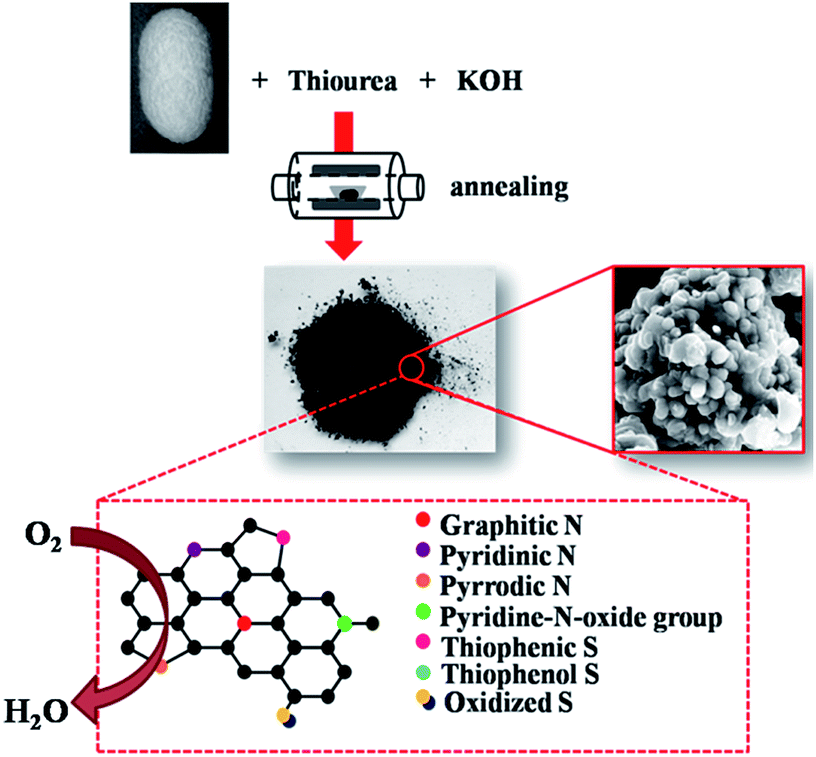 | ||
| Fig. 1 Scheme illustrating the preparation of SNC-x. | ||
Characterization
The morphology of the samples was observed by a field emission scanning electron microscope (FESEM, Hitachi S-4800, Japan) and JEM-2100HR high-resolution transmission electron microscope (HR-TEM). The Brunauer–Emmett–Teller (BET) surface areas (SBET) of samples were identified from nitrogen adsorption data recorded using a Micromeritics ASAP 2020 nitrogen adsorption apparatus. The thermal gravimetric-mass analysis were carried out by TG/DSCMS, Netzsch STA 449F3 equipment, in which samples were heated from 100 to 900 °C with a ramp rate of 10 °C min−1 in Ar atmosphere. The Fourier transform infrared spectroscopy (FTIR) analysis was recorded on a Nicolet Avatar 360 spectrophotometer as KBr pellets (USA). The Raman spectra were collected on a LabRAMII Raman spectrometer (Horiba JobinYvon, France) using a 488 nm, 50× objective, 1.3 mV laser intensity, air-cooled Ar+ laser. The X-ray photoelectron spectroscopy (XPS) spectra were operated using a VG ESCALAB MKΠ instrument (Al Kα excitation, UK).Electrochemical measurement
The electrochemical measurements were performed on a computer-controlled working station (CHI 660e, Chenhua, China) in a three-electrode electrochemical cell. The glassy carbon electrode (GCE, 7.065 mm2) or rotating ring disk electrode (RRDE-3A, ALS, Japan, 12.56 mm2) loaded with catalyst was used as the working electrode. A Pt wire and saturated calomel electrode (SCE) were employed as the counter electrode and the reference electrode, respectively. All the experiments were carried out at room temperature. The as-prepared catalysts were transferred onto the GCE or RRDE as follows: at the inception, the 5 mg catalysts were all well dispersed in the mixed solution (1 mL) of water, isopropanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and 40 μL Nafion (5 wt%) at a concentration of 5 g L−1 by sonication. The catalyst loading for CV and LSV tests are 0.354 and 0.199 mg cm−2, respectively.
:1 v/v) and 40 μL Nafion (5 wt%) at a concentration of 5 g L−1 by sonication. The catalyst loading for CV and LSV tests are 0.354 and 0.199 mg cm−2, respectively.
The cyclic voltammograms (CVs) of all samples were performed in O2-saturated and N2-saturated 0.1 M KOH electrolyte with a scan rate of 100 mV s−1. The linear sweep voltammetry curves (LSVs) of all the samples were collected with a rotating speed of 1600 rpm on a RRDE with a scan rate of 10 mV s−1. Especially, the SNC-800 catalyst was performed on the RRDE at different rotating speeds (400, 600, 900, 1200 and 1600 rpm) to investigate the ORR mechanism and primary electrocatalytic process. Methanol tolerance was examined by chronoamperometric method. For all the tests performed in O2-saturated electrolyte, O2 was bubbled into the electrolyte to reach O2 saturation during the process. The procedure for the test performed in N2-saturated electrolyte is similar except replacing O2 with N2.
The transferred electron number (n) per O2 molecule was determined according to the Koutecky–Levich (K–L) equation (eqn (1)):
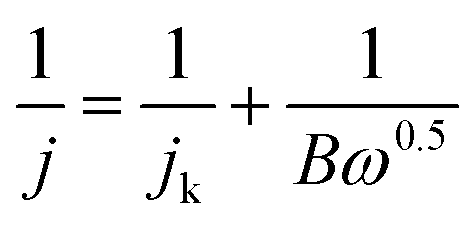 | (1) |
| B = 0.2n | (2) |
485 C mol−1); DO2 is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1); ν is the kinematic viscosity of the KOH solution (0.01 cm2 s−1) and CO2 is the concentration of dissolved O2 (1.2 × 10−6 mol cm−3).
Results and discussion
Firstly, the typical morphology of the as-prepared SNC-800 samples was characterized by means of FESEM. In Fig. 2a and b, the spatial interconnected nanoparticles around 200–500 nm diameter was observed. Interestingly, the nanoparticles with interconnected pores constructed branches of dichasial architecture, which is similar to the Astridia velutina (Fig. 2c). The cramped and loose aggregation provide numerous hierarchical pores. We suppose this unique architecture could provide abundant active sites and dramatically facilitate the transportation of reactants to the active sites, thus exceedingly ensuring the enhanced ORR activity. | ||
| Fig. 2 (a and b) SEM images of SNC-800 and (c) photograph of the Astridia velutina. | ||
As seen in Fig. 3, TEM images and BET analysis were performed. Some mesoporous channels were observed in Fig. 3a and irregular hole were signed by red circle in Fig. 3b. In addition, N2 adsorption–desorption isotherms of SNC-800 sample show a type IV curve, indicating both micro- and mesoporosity (Fig. 3c). And BET analysis reveals the Fe–N–C-800 possess large surface area of 377.0 m3 g−1 and the pore size distributions are mainly 2.7 nm (Fig. 3d). And the micropores are most around 1 nm. The results confirm the hierarchical pore structure of SNC-800.
 | ||
| Fig. 3 (a) TGA and (b) MS curves of the mixture of silk cocoon, thiourea and KOH in Ar, (c) FTIR and (d) Raman spectra of different SNC-x samples. | ||
Fig. 4a gives the TGA curve of the KOH/silk cocoon/thiourea mixture in Ar. As seen, the mixture experienced a significant mass loss up to 800 °C. The TG-MS data shown in Fig. 3b suggest the thermal degradation firstly started around 180 °C and completed at ∼300 °C. During this temperature range, the CS2, SO2, CO2, H2O, CH4, NH3, and H2S gas were released. More precisely, at ∼210 °C, the CS2 gas was liberated. Furthermore, at ∼600 and ∼700 °C, the H2S and H2 were released, respectively. This result indicates that the carbonization and doping mainly happened below 800 °C. The mass loss rate slumps beyond 800 °C, implying that the major reaction of the carbonized and doping were in the form of bond rearrangements and structural confirmation.32
 | ||
| Fig. 4 (a) TGA and (b) MS curves of the mixture of silk cocoon, thiourea and KOH in Ar, (c) FTIR and (d) Raman spectra of different SNC-x samples. | ||
To analyze the structure and composition, FTIR spectra were performed (Fig. 4c). As seen, for SNC-700 and SNC-800, the peaks around 1580 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and 1270 cm−1 (C–N) were obvious.33 The broad bands in the region of 3000–3700 cm−1 can be ascribed to the adsorbed H2O molecules and N–H vibration.34 While for the SNC-900, only peaks attributed to C–N (1000 cm−1) and CN (1580 cm−1) is noticed. The reason may be that with the increment of temperature, the content of heterogeneous elements decreased.35 Moreover, Raman is an effective tool for offering detailed information concerning the microstructure feature of carbon materials.36,37 Here, the samples synthesized at different temperatures were characterized by Raman. In Fig. 4d, the D band and G band were located near 1340 and 1580 cm−1, respectively. As known, the G band is related with all sp2-bonded structure, such as C–C, S–C and N–C, etc. While the D band arises from the sp3 defect sites.38 Generally, ID/IG is calculated as an indicator to describe the defects level. With the temperature increasing from 700 to 900 °C, the ID/IG ratio of SNC-x decreased from 3.61 to 1.24. We suppose the decline is mainly due to the following reasons. Firstly, the graphitic degree of SNC-x improved with the treat temperature. Secondly, the unceasingly elevated temperature usually lead to the decrease of the amount of dopant (S + N), which will be confirmed by the following XPS results.
N) and 1270 cm−1 (C–N) were obvious.33 The broad bands in the region of 3000–3700 cm−1 can be ascribed to the adsorbed H2O molecules and N–H vibration.34 While for the SNC-900, only peaks attributed to C–N (1000 cm−1) and CN (1580 cm−1) is noticed. The reason may be that with the increment of temperature, the content of heterogeneous elements decreased.35 Moreover, Raman is an effective tool for offering detailed information concerning the microstructure feature of carbon materials.36,37 Here, the samples synthesized at different temperatures were characterized by Raman. In Fig. 4d, the D band and G band were located near 1340 and 1580 cm−1, respectively. As known, the G band is related with all sp2-bonded structure, such as C–C, S–C and N–C, etc. While the D band arises from the sp3 defect sites.38 Generally, ID/IG is calculated as an indicator to describe the defects level. With the temperature increasing from 700 to 900 °C, the ID/IG ratio of SNC-x decreased from 3.61 to 1.24. We suppose the decline is mainly due to the following reasons. Firstly, the graphitic degree of SNC-x improved with the treat temperature. Secondly, the unceasingly elevated temperature usually lead to the decrease of the amount of dopant (S + N), which will be confirmed by the following XPS results.
The doping level and chemical environment of the doped atoms were investigated by XPS. In Fig. 5a, the XPS survey spectra of SNC-x shows peaks for O 1s (∼533.0 eV), C 1s (∼284.1 eV), N 1s (∼398.0 eV) and two S peaks (∼104.0 and ∼152.0 eV), confirming the successful doping of S and N atoms. The amounts of N and S elements in the co-doped SNC-800 were calculated to be 9.75 and 1.79 at%, respectively. The contents of N and S in the SNC-x samples were plotted against the annealing temperature (Fig. 5b). As seen, the content of N in SNC-x decreased with the increase of the annealing temperature. In contrast, the content of S increased with the increasement of the annealing temperature till 900 °C.
 | ||
| Fig. 5 (a) XPS survey of SNC-x, (b) atomic percentage of N and S with the treating temperature, (c–e) C 1s, N 1s and S 2p spectra of SNC-800. | ||
In Fig. 5c, the C 1s spectrum can be deconvoluted into three species, namely CC sp2 (284.59 eV), sp3 hybridized carbon (285.71 eV)39 and –O–CO (288.64 eV), which could result in enhanced hydrophilicity and thus enable aqueous electrolyte well infiltrated in the catalysts.40 As Fig. 5d depicted, the N 1s peak could be fitted by the four peaks at ∼397.99 (25.16%), 399.87 (34.24%), 400.75 (29.17%) and 402.50 eV (11.43%), corresponding to the pyridinic-N, pyrrodic-N, graphitic-N and pyridine-N-oxide groups, respectively.29 Among these four N functionalities, pyridinic-N and graphitic-N are believed to be highly beneficial for ORR.41 Besides, it is reported that pyridinic-N and pyrrolic-N are able to promote the incorporation of sulfur atoms to oxygen functional groups.42,43 The incorporation of sulfur into SNC-x is proved by the emergence of S 2p peaks (Fig. 5e). More precisely, those two main contributions with binding energies around 163.46 (37.84%) and 168.49 eV (44.28%) indicate that sulfur prevailingly resided in C–Sn–C (n = 1 or 2) bonds and oxidized (–SOn–) sulfur moieties. The peak at 164.58 eV (14.18%) could be attributed to the conjugated –CS– bond (thiophenic S),23 which is believed to ensure the carbon substrate positively charged.44 A minor peak at 161.76 eV (3.7%) was also detected, implying the existence of reduced sulfur (–SH). It is also worth noting that the largely bonded –SOn– moiety increases the hydrophilicity of the SNC-x samples. Thus electrolyte and dissolved oxygen could transfer to the active sites more effectively and hydroxide ions could move away from the active sites quickly to minimize electrode polarization.38
CVs were used to study the ORR performance of the as-prepared SNC-800 sample. As shown in Fig. 6a and S1,† the peak at 0.68 V were observed in O2-saturated 0.1 M KOH solution, indicating the ORR occurring on SNC-800. However, in N2-saturated solution, no peak was noticed. To gain a deeper insight into the ORR activity, LSVs of SNC-x were performed on a RDE in an O2-saturated electrolyte (Fig. 6b). SNC-800 displays more positive onset potential (0.853 V) and half-wave potential (0.717 V) as well as higher diffusion current density (4.5 mA cm−2 at 0 V), suggesting its best ORR activity among all samples. Fig. 6c displays the RDE voltammograms of SNC-800 at different rpm. Remarkably, the ORR onset potential of SNC-800 is slightly 0.1 V inferior to that of Pt/C (Fig. 6d) under a same catalyst loading of 0.24 mg cm−2. For a comparison, LSV curves of Pt/C at different rotating speed were also listed in Fig. S2.† Besides, Tafel plots of SNC-800 (Fig. S3†) derived from the corresponding LSV curves at 1600 rpm was 90 mV dec−1, close to that of Pt/C (80 mV dec−1), which suggests that SNC-800 has efficient kinetic process. In addition, SNC-800 exhibits comparable ORR activity with many other metal-free electrocatalysts, as shown in Table S1.† Along with the morphology analysis and XPS study above, we could thus reasonably deduce that the improved ORR performance of SNC-800 originates from its considerable active sites and fast electrolyte/oxygen diffusion, on account of the simultaneous introduction of S, N element and synergetic effect of Astridia velutina-like micro/mesoporous structure.
 | ||
| Fig. 6 (a) CVs of SNC-800, (b) LSVs at 1600 rpm of SNC-x samples, (c) LSVs at different rotating speed of SNC-800, (d) LSVs patterns of SNC-800 and Pt/C at 1600 rpm in an O2-saturated 0.1 M KOH solution. | ||
The K–L plots at different electrode potentials exhibit good linearity (Fig. 7a inset), and the n value for SNC-800 was calculated to be 3.83–4.17 at the potential ranging from 0.29–0.49 V (Fig. 7a), suggesting the highly desired 4e− pathway reduction for ORR on the SNC-800 electrode. Electron transfer numbers at 0.39 V (vs. RHE) of different samples are plotted against the annealing temperature (600–1000 °C), as shown in Fig. 7b. The n is calculated to be 1.34 (SNC-600), 3.81 (SNC-700), 3.95 (SNC-800), 3.16 (SNC-900) and 2.22 (SNC-1000). The 4e− pathway on the SNC-800 electrode indicates a very high ORR efficiency.
 | ||
| Fig. 7 (a) Electron transfer numbers and the corresponding K–L plots (inset) of SNC-800, (b) electron transfer numbers at 0.39 V (vs. RHE) of different samples, (c) chronoamperometric response of SNC-800 and Pt/C with methanol in O2-saturated 0.1 M KOH solution. | ||
The methanol tolerance of SNC-800 was also tested (Fig. 7c). The chronoamperometric response does not show substantial fluctuation and recovery within about 50 s after methanol injection, indicating its good tolerance to methanol. However, in stark contrast, the current density of commercial Pt/C catalyst shift from negative to positive, suggesting the dominant reaction in the catalyst system change from ORR to methanol oxidation reaction.
Conclusions
We prepared micro–mesoporous S, N-codoped SNC-x via one-step thermal treatment of silk cocoon, thiourea and KOH, creating unique architectures with abundant ORR active sites and hierarchical porous structure for fast reactant transportation. The remarkable ORR activity should be mainly attributed to the incorporation of S, N co-doping and unique pore structure. Considering that commercially available silk cocoon is relatively inexpensive and the simple synthetic approach is applicable for mass production, the metal-free SNC-800 could be a promising alternative for the new generation of cost-effective catalysts in energy and environment related fields.Acknowledgements
The work was financially supported by National Natural Science Foundation of China (51203182), Foundation for the Author of Excellent Doctoral Dissertation of Hunan Province (YB2014B004), Key Laboratory of Advanced Textile Materials and Manufacturing Technology (Zhejiang Sci-Tech University), Ministry of Education (2015001), Research Project of NUDT (ZK16-03-32), Key Laboratory of Lightweight and Reliability Technology for Engineering Vehicle, College of Hunan Province (2016kfjj01). We thank Hong Wang and Tengyuan Wang for help in experiment. Also, we thank Qi Shi and Qichen Wang for helpful discussions.References
- X. P. Gao and H. X. Yang, Energy Environ. Sci., 2010, 3, 174 CAS.
- T. Cao, D. Wang, J. Zhang, C. Cao and Y. Li, Chem.–Eur. J., 2015, 21, 14022 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294 CrossRef CAS PubMed.
- N. Wu, Y. D. Wang, Y. P. Lei, B. Wang, C. Han, Y. Z. Gou, Q. Shi and D. Fang, Sci. Rep., 2015, 5, 17396 CrossRef CAS PubMed.
- J. Li, G. Wang, J. Wang, S. Mao, M. Wei, F. Yang, L. Yu and X. Bao, Nano Res., 2014, 7, 1519 CrossRef CAS.
- X. J. Zhou, J. L. Qiao, L. Yang and J. J. Zhang, Adv. Energy Mater., 2014, 4, 1301523 CrossRef.
- G. Zhang, Y. Xu, L. Wang, J. Wang, Y. Kuang and X. Sun, Sci. China Mater., 2015, 58, 534 CrossRef.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634 CrossRef CAS.
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J. B. Baek and L. M. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209 CrossRef CAS PubMed.
- S. Ci, P. Cai, Z. Wen and J. Li, Sci. China Mater., 2015, 58, 496 CrossRef.
- R. Ma, X. Ren, B. Y. Xia, Y. Zhou, C. Sun, Q. Liu, J. Liu and J. Wang, Nano Res., 2016, 9, 808–819 CrossRef CAS.
- S. Wang, E. Iyyamperumal, A. Roy, Y. Xue, D. Yu and L. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756 CrossRef CAS PubMed.
- Z. Liu, G. Zhang, Z. Lu, X. Jin, Z. Chang and X. Sun, Nano Res., 2013, 6, 293 CrossRef CAS.
- D. S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. S. Yu, J. Am. Chem. Soc., 2012, 134, 16127 CrossRef CAS PubMed.
- Q. Shi, Y. D. Wang, Z. M. Wang, Y. P. Lei, B. Wang, N. Wu, C. Han, S. Xie and Y. Z. Gou, Nano Res., 2016, 9, 317–328 CrossRef CAS.
- Q. Shi, Y. P. Lei, Y. D. Wang and Z. M. Wang, J. Inorg. Mater., 2016, 31, 351–357 CrossRef.
- Y. P. Lei, Q. Shi, C. Han, B. Wang, N. Wu, H. Wang and Y. D. Wang, Nano Res., 2016, 9, 2498–2509 CrossRef.
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339 CrossRef CAS PubMed.
- Q. Shi, Y. P. Lei, Y. D. Wang, H. P. Wang, L. H. Jiang, H. L. Yuan, D. Fang, B. Wang, N. Wu and Y. Z. Gou, Curr. Appl. Phys., 2015, 15, 1606 CrossRef.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084 CrossRef CAS PubMed.
- Y. Su, Y. Zhang, X. Zhuang, S. Li, D. Wu, F. Zhang and X. Feng, Carbon, 2013, 62, 296 CrossRef CAS.
- Y. Huang, P. Wu, Y. Wang, W. Wang, D. Yuan and J. Yao, J. Mater. Chem. A, 2014, 2, 19765 CAS.
- S. Gao, K. Geng, H. Liu, X. Wei, M. Zhang, P. Wang and J. Wang, Energy Environ. Sci., 2015, 8, 221 CAS.
- H. Liu, Y. Cao, F. Wang and Y. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 819–825 CAS.
- K. N. Chaudhari, M. Y. Song and J. S. Yu, Small, 2014, 10, 2625 CrossRef CAS PubMed.
- F. Pan, J. Jin, X. Fu, Q. Liu and J. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 11108 CAS.
- Y. R. Liang, D. C. Wu and R. W. Fu, Sci. Rep., 2013, 3, 01119 Search PubMed.
- Y. L. Cheng, L. Huang, X. Xiao, B. Yao, L. Y. Yuan, T. Q. Li, Z. M. Hu, B. Wang, J. Wan and J. Zhou, Nano Energy, 2015, 15, 66 CrossRef CAS.
- B. Zhang, J. Q. Huang and J. K. Kim, Adv. Funct. Mater., 2015, 25, 5222 CrossRef CAS.
- Y. Li, H. Zhang, Y. Wang, P. Liu, H. Zhang, X. Yao, D. Wang, Z. Tang and H. Zhao, Energy Environ. Sci., 2014, 7, 3720 CAS.
- W. Shen, S. Zhang, Y. He, J. Li and W. Fan, J. Mater. Chem., 2011, 21, 14036 RSC.
- J. Hong, X. Xia, Y. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006 RSC.
- X. Wang, J. Wang, D. Wang, S. Dou, Z. Ma, J. Wu, L. Tao, A. Shen, C. Ouyang, Q. Liu and S. Wang, Chem. Commun., 2014, 50, 4839 RSC.
- Y. P. Lei, Y. D. Wang and Y. C. Song, Ceram. Int., 2013, 39, 6847 CrossRef CAS.
- Y. P. Lei, Y. D. Wang, Y. C. Song and C. Deng, Ceram. Interfaces, 2011, 37, 3005 CrossRef CAS.
- S. Dou, A. L. Shen, L. Tao and S. Y. Wang, Chem. Commun., 2014, 50, 10672 RSC.
- F. Razmjooie, K. P. Singh, M. Y. Song and J. S. Yu, Carbon, 2014, 78, 257 CrossRef.
- J. S. Lee, G. S. Park, S. T. Kim, M. Liu and J. Cho, Angew. Chem., Int. Ed., 2013, 52, 1026 CrossRef CAS PubMed.
- T. Cao, D. S. Wang, J. T. Zhang, C. B. Cao and Y. D. Li, Chem.–Eur. J., 2015, 21, 14022 CrossRef CAS PubMed.
- J. X. Song, T. Xu, M. L. Gordin, P. Y. Zhu, D. P. Lv, Y. B. Jiang, Y. S. Chen, Y. H. Duan and D. H. Wang, Adv. Funct. Mater., 2014, 24, 1243 CrossRef CAS.
- P. Y. Zhu, J. X. Song, D. P. Lv, D. H. Wang, C. Jaye, D. A. Fischer, T. P. Wu and Y. S. Chen, J. Phys. Chem. C, 2014, 118, 7765 CAS.
- F. Wu, J. Li, Y. F. Tian, Y. F. Su, J. Wang, W. Yang, N. Li, S. Chen and L. Y. Bao, Sci. Rep., 2015, 5, 13340 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06664j |
| This journal is © The Royal Society of Chemistry 2016 |