
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monoclonal antibody capture from cell culture supernatants using epitope imprinted macroporous membranes
Sebastian
Schwark
c,
Wei
Sun
a,
Jörg
Stute
d,
Dirk
Lütkemeyer
d,
Mathias
Ulbricht
*c and
Börje
Sellergren
*ab
aFaculty of Chemistry, Technical University of Dortmund, Germany
bDepartment of Biomedical Sciences, Faculty of Health and Society, Malmö University, Malmö, Sweden. E-mail: borje.sellergren@mah.se
cLehrstuhl für Technische Chemie II, Universität Duisburg-Essen, Essen, Germany
dBibitec GmbH, Bielefeld, Germany
First published on 24th May 2016
Abstract
Epitope-imprinted membranes targeting the C-terminal fragment of the immunoglobuline G (IgG) heavy chain was developed and used for the purification of a commercial monoclonal antibody. The membranes exhibited strongly enhanced IgG affinity when compared with non-imprinted or IgG imprinted membranes reflected in binding selectivities in a protein mixture (IgG/HSA 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 w/w) of up to 40, and the elution of 95 to 100% pure IgG after washing. The dynamic binding capacity amounted to 3.9 mg mL−1 membrane volume with minor loss in performance upon repeated cleaning with alkali. The depletion of host cell proteins from a cell culture broth after production of anti-IL8 antibody using the best performing imprinted membrane under low-salt conditions reached 88% (0.7–1.2 log units) implying an effective removal of impurities from the cell culture supernatant.
:10 w/w) of up to 40, and the elution of 95 to 100% pure IgG after washing. The dynamic binding capacity amounted to 3.9 mg mL−1 membrane volume with minor loss in performance upon repeated cleaning with alkali. The depletion of host cell proteins from a cell culture broth after production of anti-IL8 antibody using the best performing imprinted membrane under low-salt conditions reached 88% (0.7–1.2 log units) implying an effective removal of impurities from the cell culture supernatant.
Introduction
Monoclonal antibodies (mAb), i.e. the use of proteins as the active pharmaceutical ingredient, are one of the most rapidly growing segments of biopharmaceuticals.1 They are currently used for the treatment of a wide spectrum of diseases, e.g. cancer, or autoimmune, cardiac and various infectious diseases. However, for mAbs presently reaching the market, therapy costs are high which can be ascribed mainly to insufficient production capacities.2 Various host cell cultures are used, but the major bottleneck can be found in the downstream processing, commonly divided into a capturing, a separation and a polishing step for purifying the mAb from the crude fermentation broth.1,3 The capture step is presently dominated by the use of protein A, a 42 kDa protein with high affinity for the Fc region of IgG. This still imperfect affinity separation suffers from high costs, poor stability, toxic leachables and harsh elution conditions. Hence, numerous attempts have been made to develop cheap, robust and easy to use alternatives for mAb capture. These comprise alternative IgG binding proteins (e.g. protein G, protein L),4 synthetic protein A mimics,5 as well as bioengineered peptides and synthetic ligands.6 With a few exceptions these approaches involve two steps: (1) optimization of the protein binder, and (2) immobilization of the binder to a suitable support. Often a time consuming optimization of ligand orientation, density and matrix is here required in order to reach adequate binding properties. One step approaches where the protein binding site and the porous separation material is formed simultaneously appear attractive in this context.5 One such approach is molecular imprinting for the preparation of robust artificial protein binders.7 In spite of numerous reports describing protein imprinted hydrogels, advances towards generic and robust imprinting techniques have been slow. One reason for this is the need for employing a low cross-linking level in order to provide a mesh size of the network large enough for the protein to penetrate. The memory effects of these gels are thus easily erased preventing repeated use. Various forms of surface imprinting and high dilution polymerization techniques have been used with promising results to address this problem.8 However, robust imprinting techniques affording materials which can compete with established bioaffinity media in terms of both affinity and capacity are still lacking. The most viable approach to produce protein recognizing molecularly imprinted polymers (MIPs) is based on the use of epitopes as templates, i.e. a short peptide corresponding to a solvent exposed, usually terminal sequence of the protein target (Fig. 1).8a,9 The appropriate sequence can be selected based on analysis of the protein crystal structure or immune-based epitope mapping while minimizing solvent induced conformational changes under imprinting versus assay conditions.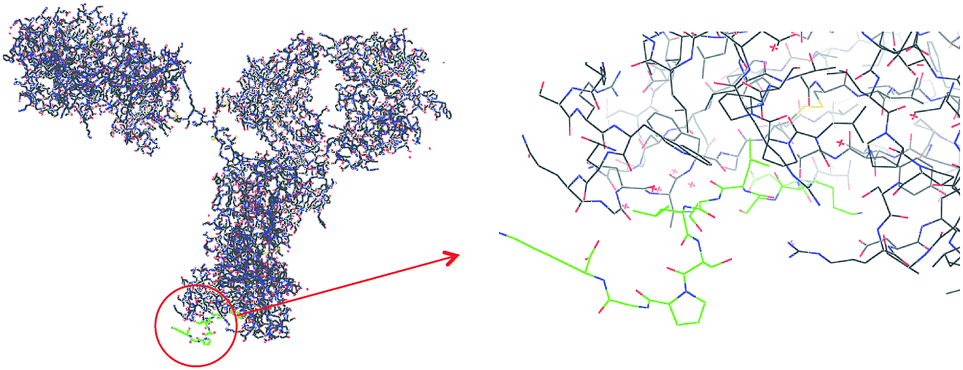 | ||
| Fig. 1 Part of the sequence of IgG heavy chain (from PDB, code 1HZH) and the synthetic peptide (QKSLSLSPGK; “T10”; labeled green) used in the study. | ||
Aiming at identifying a conserved and solvent accessible sequence common for the IgG class of monoclonal antibodies we had focused our attention on the terminal regions of the heavy chains. In a parallel study, we have found that the C-terminal decapeptide sequence displayed in Fig. 1 satisfies these criteria and we have shown that corresponding epitope-imprinted polyacrylamide gels can be optimized to selectively cross-react with commercial IgG and a monoclonal antibody.10 In an effort to practically apply this affinity principle in protein purification we have here transferred the gel format to a membrane based capture format (Fig. 2).11
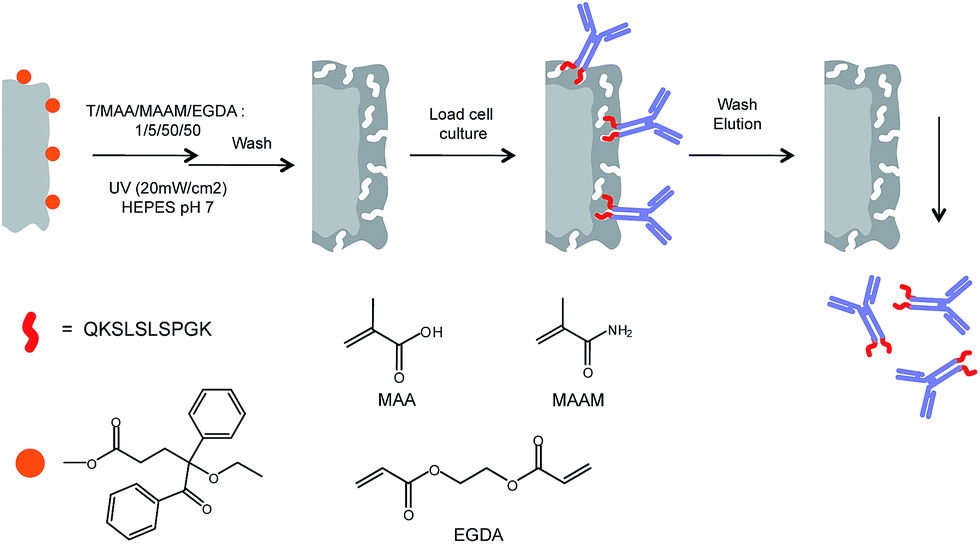 | ||
| Fig. 2 Procedure for surface-initiated graft copolymerization of antibody-imprinted hydrogel layers on cellulose membrane surfaces (T = template; here: QKSLSLSPGK; “T10”; cf.Fig. 1). | ||
Macroporous membranes endowed with chemically selective binding properties are an important addition to the well established membranes designed for size selective separations.12 Such membranes are used in membrane chromatography or affinity chromatography for various purification tasks and feature several attractive properties vis a vis packed column based separations. The main benefit is the rapid mass transfer associated with these separations which almost entirely occurs by convection through the pores, with minimal diffusion limitations. The binding capacity is hence often independent of flow rate. A convenient technique to modify membrane pore walls with uniform polymer films is the “grafting-from” technique which is based on the initiation of chain growth at the support surface.13 This has proven to be a successful strategy for generating thin film surface imprinted membranes. The grafting density or film thickness can here be easily adjusted and this has turned out to be a key parameter for optimizing selectivity, affinity and accessibility with respect to target molecules of different sizes including proteins.8f,14
We recently introduced a “grafting-from” technique relying on a covalently surface-immobilized “type I” photoinitiator,15 a benzoin ethyl ether (BEE) derivative. The technique allows a precise control of the grafting yield through the density of immobilized initiator and the UV irradiation time. It is also associated with less side reactions compared to alternative grafting-from techniques (e.g. iniferter, atom transfer radical polymerization (ATRP), reversible addition fragmentation chain transfer polymerization (RAFT)). This technique we here adapted to the grafting of epitope- and protein-imprinted polymer hydrogel films from macroporous cellulose membranes (cf.Fig. 2). The influence of preparation conditions had been evaluated and the use of the best epitope-imprinted membrane under near process, realistic conditions leads to efficient purification of a recombinant anti-interleukin antibody from cell culture supernatants.
Experimental
Methacrylamide (MAAm), methacrylic acid (MAA), ethyleneglycoldiacrylate (EGDA), benzoin ethyl ether (BEE), benzoic acid bromide, tetrahydrofuran, acetonitrile (HPLC grade) and the buffer salts were obtained from Sigma-Aldrich (Germany). Triethylamine was from Merck (Germany). The IgG (>95%) used as template for imprinting and Human Serum Albumin (HSA) were also from Sigma-Aldrich. The C-terminal epitope peptide (QKSLSLSPGK) of a minimum purity of 95% was obtained from GenScript (Piscataway, USA).The IgG used for the capturing studies was a monoclonal antibody against the cytokine interleukin-8 (anti-IL-8-mAb) produced by a CHO cell line ATCC CRL-12445 (Bibitec GmbH, Bielefeld, Germany). Prior to use, the high salt protein solution was dialyzed in a Nadir membrane tube (pore size 25–30 Å, from Roth GmbH, Germany) against a large excess of water for 2 days (3 × daily changes), followed by lyophilization.
Cellulose membranes (RC 60, nominal pore diameter 1.0 μm) from Whatman (UK) had a thickness of 73 ± 3 μm, a measured mean gas flow pore diameter of 1.2 μm and a BET surface area of 6.3 m2 g−1,15 and the characteristic pore structure is shown in Fig. 3.
 | ||
| Fig. 3 Scanning electron microscopy image of the macroporous cellulose membrane used in this study. | ||
Photoinitiator immobilization
The photoinitiator 4-ethoxy-5-oxo-4,5-diphenylpentanoic acid (BEE-COOH) was synthesized from BEE according to literature,16 then converted to the carboxylic acid bromide (BEE-COBr) and thereafter immobilized to the cellulose membranes, all as reported previously.15 In brief, a cellulose membrane (diameter 47 mm) was immersed in a solution of 50 mg BEE-COBr and 50 mg triethylamine in 3 ml dry tetrahydrofurane and reacted for 4 hours at 55 °C under shaking in gas-tightly sealed vessel. Thereafter, the membrane was washed, first with ethanol, then with water, and then again with ethanol, and ultimately dried at 50 °C. For the variation of immobilized photo-initiator density, the same protocol was used, but certain fraction of the photo-initiator derivative BEE-COBr were replaced by benzoic acid bromide.Preparation of IgG or epitope imprinted membrane adsorber
Membranes, pre-functionalized with photo-initiator (photo-initiator content 10%, 50% or 80%) as described above (diameter 47 mm) were immersed in 1 ml monomer solution placed in a Petri dish with a slightly smaller Petri dish as a cover to prevent uptake of oxygen from air and as UVA filter.15 The monomer mixture consisted of 10 mM MAA, 100 mM MAAm, and 100 mM EGDA in water. This was used for preparation of non-imprinted reference materials (NIP). For imprinting, either 2 mg of the peptide template T10 or 2 mg of the native protein (IgG) were added as template to 1 ml of that solution. After 10 min equilibration between membrane and reaction mixture, the UV irradiation was carried out in a UV cube 2000 (Hönle, Germany) for 10 or 30 minutes at an intensity of 20 mW cm−2. Thereafter, the membranes were step-wise washed with 20 ml of ethanol, 20 ml of 1 M sodium chloride solution, 20 ml of water and again 20 ml of ethanol, for 30 min in each step, and ultimately, they were dried.Membrane characterisation
Photo-initiator immobilization including variation of photo-initiator density on the cellulose surface had in detail been analyzed in a previous study;15 the BEE densities on the surface were ∼50, ∼100 and ∼380 pmol cm−2 (relative to specific surface area), respectively, for 10, 50 and 80% BEE in the solution used for initiator immobilization. The degree of grafting (DG) was determined gravimetrically from the weight of the membrane sample before and after functionalization. Here, the values were then normalized to the specific surface area of the base membrane (6.3 m2 g−1; cf. above). It had also been confirmed that the specific surface area does not change significantly due to photo-initiator immobilization. Each functionalization had been done at least 3 times, and average values with standard deviation are reported.Analysis of protein binding capacity and selectivity
The protein binding properties for the wide range of imprinted and non-imprinted membranes had been done in batch experiments in the bind–wash–elute mode. All experiments were performed at pH = 7.4, adjusted by a HEPES buffer (25 mM).For binding, one membrane (diameter 47 mm) was immersed in 3 ml protein solution (either IgG or HSA, 0.5 mg ml−1) in a polyethylene vessel (diameter 50 mm), the vessel was sealed and incubation was done for 4 hours at room temperature on a shaker (100 rpm). Then membranes were taken out and washing was done 2 times for 30 min with 10 ml buffer. Thereafter, desorption was done by immersing the membrane in 3 ml buffer with additional 1 M NaCl for 4 hours at room temperature on a shaker (100 rpm). Quantitative analysis of the protein content in the solution after binding and the eluate solution obtained after washing with buffer was done by using the BCA™ protein assay kit (Thermo Scientific) in 96 well microplates comprising also respective protein standards. 25 μl protein solution and 200 μl reagent were incubated for 30 min at 37 °C on a shaker (300 rpm) and photometric measurement at 562 nm was then done using a μQuant™ microplate spectrophotometer (BioTek Instruments).
Results are protein binding capacities after the first step (”bind”; mad) and the capacity with respect to eluted protein, i.e. after washing (”elute”; mel) which are normalized to the membrane volume (calculated from sample area and membrane thickness; cf. above). The selectivities between IgG and HSA, in the binding and in the elution step, are obtained by the ratio of these values:
| αad_IgG/HSA = mad,IgG/mad,HSA |
| αel_IgG/HSA = mel,IgG/mel,HSA |
Additional experiments on reusability were also performed by repeating the bind–wash–elute cycles with the same membrane for 3 times; in between the cycles membranes had been re-equilibrated with the binding buffer (25 mM HEPES, pH = 7.4).
For each membrane and protein, 3 independent samples were analysed and average values with standard deviation are reported.
Protein capture from binary protein mixtures
For selected imprinted membranes, the separation of IgG/HSA mixtures was also analyzed in flow-through bind–wash–elute mode. A stack of 5 membranes (diameter 32 mm) was assembled in a flow-through cell (custom made in the university workshop) and this was used as a very short column in an Äkta™ Purifier system (GE Pharmacia). A sample loop of 1 ml was used, the flow rate was always 0.25 ml min−1, and UV detection at 280 had been done. A protein mixture (1 g l−1 HSA, and 0.1 g l−1 IgG) was dissolved in 25 mM HEPES buffer (pH = 7.4). First, the membrane was equilibrated with the HEPES buffer until a stable base line had been obtained. Then, the protein solution was injected, and 10 ml HEPES buffer were used for subsequent washing. The first 4 ml were collected and subsequently analyzed by HP-SEC. Thereafter, elution was done with 4 ml HEPES buffer containing additional 1 M NaCl; the eluate was collected and subsequently also analyzed by HP-SEC (see below). Ultimately, the system was again equilibrated with low salt HEPES buffer for the next run. Up to 5 repeated evaluations of the same membrane stack, also with single protein samples, had been performed.Analysis of the protein mixtures had been done by high performance size exclusion chromatography (HP-SEC). An Äkta™ Purifier system with a Superdex 200 10/30 column (GE Healthcare), a mobile phase consisting of 80% PBS buffer and 20% acetonitrile, a flow rate of 0.25 ml min−1 and UV detection at 280 nm were used.
Preparation of cell culture supernatant
For the project, the recombinant CHO cell line DP – 12 (clone # 1934, ATCC Nr. CRL – 12445) was used. This cell line produces a monoclonal humanized antibody (IgG1) directed against human interleukin 8 (IL-8). For the culturing of the cells a proprietary, serum-free and protein-poor medium was used. For inoculation of 100 l bioreactor the cells were cultivated over 4 stages (20 ml to 10 l) and for a total of 26 days. Until the end of cultivation a cell density of 1.1 × 107 cells per ml was reached. The viability was always >90%. At the end of cultivation an antibody titer of 50 mg l−1 was achieved. After completion of the cultivation run in the 100 l bioreactor, the cell removal was carried out. A pre-treatment of the culture solution was done by sedimentation at 5 °C for 15 hours. During this time, the antibody concentration increased to 63 mg l−1. Finally, cellular debris and organelles were removed by centrifugation at 10 °C and microfiltration.Protein capture from cell culture supernatants using protein A affinity column
To purify the anti-IL-8 antibody, the material “MabSelect Sure” (GE Healthcare) was used. The protein A based affinity resin binds specifically to antibodies of the IgG class, which leads to a high purity of the reclaimed product. The volume of the used chromatographic column was 151 ml. This column could process up to 30 l cell-free culture supernatant within one working day. A total of 90 l culture supernatant (cf. above) was purified in five cycles. The culture supernatant was loaded directly onto the affinity column equilibrated with binding buffer (50 mM Tris/HCl, 150 mM NaCl pH 8.0; residence time 1.8 min). After washing with binding buffer (15 column volumes), the elution of the antibody was conducted by lowering the pH (50 mM Tris/HCl, 50 mM Na-citrate, 150 mM NaCl pH 2.5; residence time 3.4 min). The eluate was neutralized directly with 500 mM Tris/HCl, 150 mM NaCl pH 8.5. The yields of protein A chromatography were always >90%. The efficiency of purification was analyzed by silver-stained SDS-PAGE under reduced and non-reduced conditions. The depletion of host cell proteins (HCPs) was also determined by a commercial available CHO HCP ELISA (catalog # CM015; Cygnus Technologies).Dynamic binding capacity and regeneration stability of protein A affinity column
Stability and constant performance of the “Mabselect Sure” affinity column was demonstrated over 40 cycles. A cleaning-in-place procedure with 0.5 M NaOH was conducted every 8 cycles. The dynamic binding capacities of used and unused resins were determined by breakthrough curves. After fractionated loading of the protein A column with cell culture supernatant, the different fractions of flow-through were analyzed with respect to anti-IL-8 antibody content. The dynamic binding capacity refers to the amount of loaded protein at 10% breakthrough.Protein capture from cell culture supernatants using imprinted membrane adsorber
The membranes were used in six layers having a total area of 96 cm2 (bed volume 0.8 ml) in a filter holder. Protein A purified anti-IL-8-antibody prediluted with binding buffer (25 mM HEPES, pH = 7; conductivity < 3 mS cm−1) was loaded onto the membranes to reach a total load of 0.6–0.9 mg ml−1 membrane. The residence time was 5 minutes. Thereafter the membranes were washed with binding buffer and then the antibody was eluted with 1 M NaCl. The yield of recovered IgG was determined for 5 repeated bind–wash–elute cycles, with intermediate cleaning of the membranes with 0.1 M NaOH. The dynamic binding capacity was determined as the amount of loaded protein at 10% breakthrough.Results and discussion
Preparation and characterisation of imprinted macroporous membrane adsorbers
As a first step we decided to compare epitope imprinting with the classical protein imprinting approach relying on the use of the native protein target as template. The template in the former case consisted of a decapeptide corresponding to the C-terminal sequence of the IgG heavy chain (Mw = 1044 Da, pI = 10, gravy index = −0.81). In terms of template size, this contrasted with the nearly 150 times larger template used in the protein imprinting approach (Mw = 150 kDa, pI ≈ 7). The templates were imprinted using the MAA as acidic functional monomer, chosen for its ability to interact ionically as well as via hydrogen bonding with the positively charged templates. Additional hydrogen bond stabilization we anticipated would be provided by the primary amide monomer MAAm. The hydrophobic cross-linker EGDA we anticipated would offer complementary apolar interactions with the hydrophobic segments of the template apart from stabilizing the three dimensional arrangement of binding groups.Previous studies have established grafting density as a key parameter in controlling selectivity, affinity and accessibility of imprinted surface grafted polymer films. Hence, the optimum film thickness in terms of recognition related properties typically scales with the template size.8f,17 The previously established grafting method based on a covalently immobilized “type I” photo-initiator had been proven to be very well suited for adjusting the initiator and, hence, polymer grafting density.15 UV irradiation time is another parameter which has an effect on the effective grafted film thickness. However, given that molecular imprinting typically involves formation of cross-linked terpolymers or higher order polymers this effect can only be assessed empirically. A series of membranes were therefore prepared from identical monomer mixtures while varying the surface coverage with initiator and irradiation time. The resulting materials were first characterised with respect to the degree of grafting (Fig. 4A). Considering first the nonimprinted materials (NIP), both the time of UV irradiation and the density of immobilized initiator can be used to systematically control the degree of grafting (DG). The increase in DG with UV irradiation time was nonlinear and appeared to level off after 10 min irradiation; this can possibly be ascribed to irreversible conversion of the limited amount of initiator on the surface. The DG was about 2 times higher for the membranes prepared using 50% nominal initiator coverage compared to those prepared using 10% coverage. This is in agreement with the difference in measured coverage of the initiator groups (100 vs. 50 pmol cm−2; cf. Experimental section) while at higher initiator densities (380 pmol cm−2 for 80% nominal coverage) grafting seems to be impeded possibly due to initiator or primary chain radical recombination. For short irradiation times, the addition of IgG seems to interfere with the formation of the cross-linked polymer hydrogel film, while for longer irradiation times the DG values for MIP and NIP were identical within the range of error. The addition of the epitope template T10 led to a slightly higher DG. After the grafting step, the templates were removed by subjecting the membranes to successive washes with ethanol, 1 M NaCl, ethanol and water.
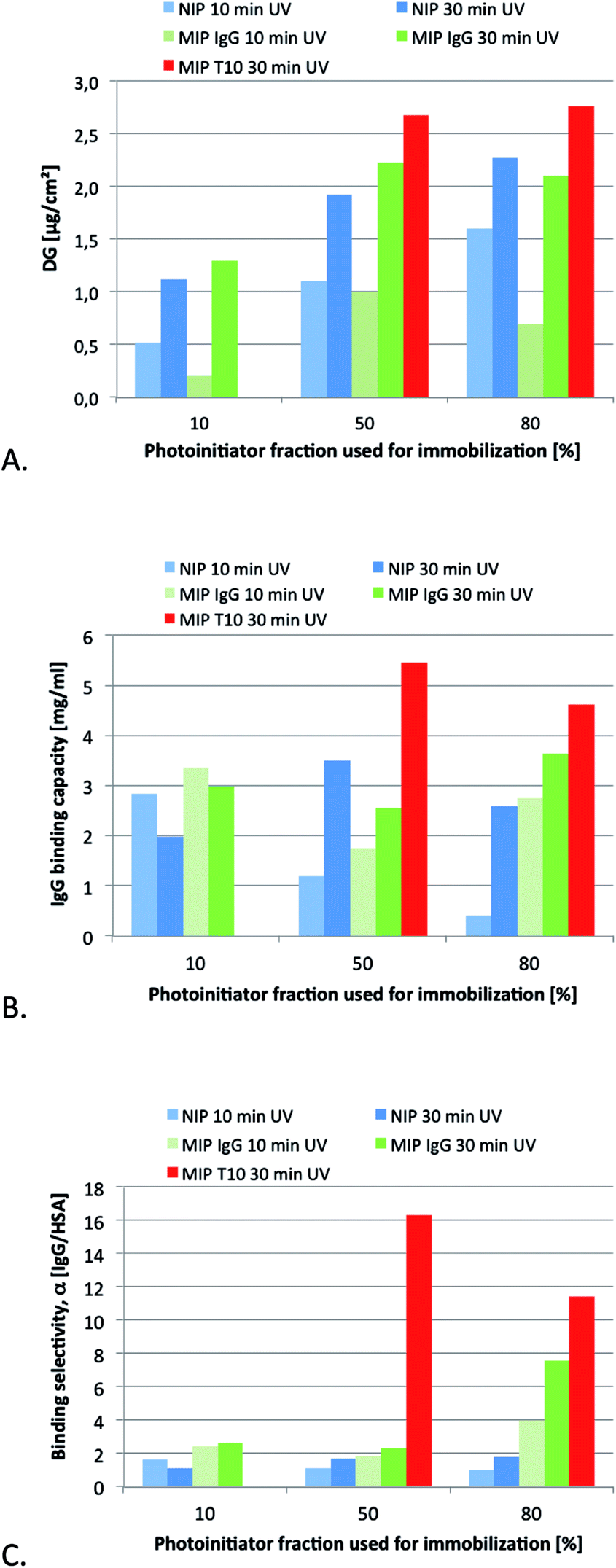 | ||
| Fig. 4 (A) Degree of grafting versus UV irradiation time and initiator density for non-imprinted reference materials (NIP) and imprinted membranes (MIP) grafted using the native IgG or the peptide epitope template (T10). (B) Amount of IgG adsorbed and (C) IgG/HSA selectivity of the membranes from single protein experiments (protein concentration 0.5 g L−1 in HEPES buffer, 25 mM, pH = 7.4) after washing with HEPES buffer and elution with HEPES buffer containing 1 M NaCl. Maximum standard deviations were ±15% for DG values and ±10% for protein binding capacities (cf. Experimental part). | ||
Because of the higher monomer conversion and DG values, we used the extended grafting time of 30 min to produce membranes for a more detailed characterisation of the binding properties. Single protein binding experiments were first performed in order to evaluate binding capacity and selectivity of the materials. The results are displayed in Fig. 4B and C.
Overall, the difference in IgG capacities between the different membranes was not very large. With the exception for the NIP 10% membrane, all values are in the range of monolayer capacity for this membrane type (estimated from specific surface area and size of the proteins ∼ 5 mg ml−1). Compared to the low-protein binding cellulose support membrane the values are significantly higher which can be related to the reactively high fraction of hydrophobic cross-linker (EGDA) used for the preparation of the grafted polymer layers.
However, a striking difference can be seen with respect to the protein selectivity. All NIP membranes and MIP membranes prepared with IgG as template and up to 50% nominal initiator coverage featured only a small difference in the uptake of the two proteins HSA and IgG. This stands in sharp contrast to the performance of the membranes prepared with the epitope template. Here the binding of IgG exceeded that of HSA more than 11 fold. It should be noted that these results were obtained in a noncompetitive binding experiment. Interestingly, even higher protein selectivities were obtained when carrying out the binding experiment under competitive conditions (see below). It is also noteworthy that membranes prepared using the protein template (IgG) in combination with the highest photo-initiator density displayed enhanced protein selectivities. This agrees qualitatively with results from our previous studies on surface grafted IgG-imprinted membrane adsorbers where also the entire protein had been used as template.8f It is interesting to note that maximum uptake and selectivity was in this case obtained for membranes prepared using the highest initiator density whereas imprinting of the smaller peptide template displayed best results in combination with the intermediate initiator density. These slightly contrasting behaviour is possibly related to different film thickness of the membranes although Fig. 4A indicates no differences between the membranes. The importance of carefully matching the thickness of grafted films to the size of the template has been amply demonstrated in the literature.8f,17
Downstream processing relies not only on specific capture but also on the ability to mildly release the target protein in functional form. It was therefore gratifying to note that the protein could be quantitatively released from some membranes under high salt conditions. This supports a retention mechanism dominated by electrostatic interactions which is in complete agreement with protein–membrane interactions involving predominantly the solvent exposed charged C-terminal fragment.
Indeed, mass balance calculations revealed that 100% of loaded IgG could be eluted from T10-imprinted membranes prepared using 50% initiator coverage whereas the recovery dropped somewhat for membranes prepared using T10-imprinted membranes containing higher initiator coverage; i.e. a higher grafting density compromised in this case both capacity and selectivity. In view of the promising performance, especially of the former epitope imprinted membrane (high binding capacity: 5 mg ml−1; high selectivity: α = 16; high recovery: 100%), the two epitope imprinted membranes were subjected to further more detailed characterisation.
An epitope-imprinted membrane adsorber for antibody capture
The re-usability of the two types of T10-imprinted membranes was first tested in both batch and flow-through modes. Up to 5 subsequent bind–wash–elute cycles were performed on a minimum of 3 different membranes of each type, from single protein (IgG or HSA) solutions or binary protein mixtures (IgG/HSA). After protein elution with high salt buffer a simple regeneration by equilibration with low salt binding buffer was sufficient for reconditioning the membrane for a subsequent capture experiment. No significant change of binding capacity could be observed. Hence, the cross-linked polymer film is robust enough for repeated use.The separation performance for a model protein mixture, with HSA in 10 fold excess compared to IgG, had been studied under chromatographic flow-through conditions; the results are shown in Table 1. The IgG binding capacities were somewhat lower compared to the single protein experiments; that could be caused by the lower IgG concentration (0.1 vs. 0.5 mg l−1) used in the mixture study. The binding of HSA was much lower, resulting in high selectivity values (almost 40 after binding step). The fact that the selectivity under these competitive conditions was much higher compared to the single protein experiments confirms that the imprinted sites have indeed a much higher affinity for the target protein compared to a non related, smaller protein. In the eluted samples only IgG could be detected; the estimated selectivity of >300 results from the estimated detection limit for HSA under the used conditions (with size exclusion chromatography as reference analytics). Moreover, for the MIP T10 50% membrane, IgG recovery had been 100% while 95% had been found for the MIP T10 80% membrane. This small difference is analogous to the findings after single protein binding studies (cf. above), again supporting the slightly superior affinity distribution of the membrane prepared at lower grafting density. With respect to capacity, both membrane types are equal. Based on these very promising results with model proteins, the MIP T10 50% MIP membrane was further evaluated in studies of IgG capture from fermentation broths.
| Protein | MIP (50% initiator) | MIP (80% initiator) | |||
|---|---|---|---|---|---|
| Q (mg ml−1) | α IgG/HSA | Q (mg ml−1) | α IgG/HSA | ||
| Binding | IgG | 3.6 ± 0.2 | 36 | 3.9 ± 0.3 | 39 |
| HSA | 0.1 ± 0.03 | 0.1 ± 0.03 | |||
| Elution | IgG | 3.6 ± 0.2 | > 300 | 3.7 ± 0.3 | > 300 |
| HSA | n.d. | n.d. | |||
| IgG recovery | 100% | 95% | |||
Antibody capture by protein A affinity chromatography
As a benchmark method for the capture of the anti-interleukine (IL-8) antibody we chose a commercial protein A affinity column since this is currently the “gold standard” for IgG capture. Protein A interacts specifically with the Fc region of IgG and can be used to capture and release the protein from cell culture broth leading to a high purity of the recovered product. The aim of the benchmark test was the collection of reference data in terms of capacity, number of cycles, NaOH stability and depletion of host cell proteins (HCP). A total of 90 l of the culture supernatant were purified in five cycles. The culture supernatant was loaded directly onto the affinity column after equilibration with binding buffer. After washing with binding buffer, the elution of the antibody was carried out by lowering the pH. The yields were >90% of purified antibodies. The stability and consistent performance of the capture could be shown for about 40 cycles. After every 8 cycles a cleaning-in-place procedure (CIP) with 0.5 M sodium hydroxide was performed. Fig. 5 shows typical breakthrough curves of the affinity column after loading of culture supernatant. The dynamic capacity, defined as the load at a 10% product breakthrough, of a fresh virgin resin was 44 mg IgG per ml of gel. After a total of 40 cycles and 5 CIP procedures, the capacity dropped to 25 mg IgG per ml of gel. This may be related to the partial deactivation of the bioaffinity ligand protein A.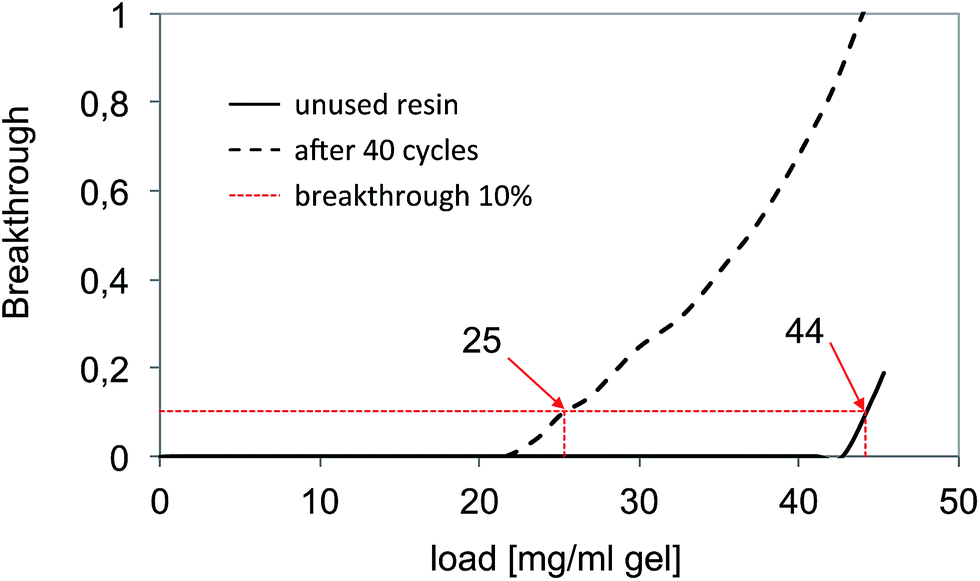 | ||
| Fig. 5 Determination of the dynamic binding capacity of the reference bioaffinity material “MabSelect Sure”. | ||
The depletion of host cell proteins determined by ELISA was found to be 3 log steps. As revealed by Fig. 6A, the excellent HCP depletion using this procedure is clearly demonstrated also by the complementary method SDS-PAGE.
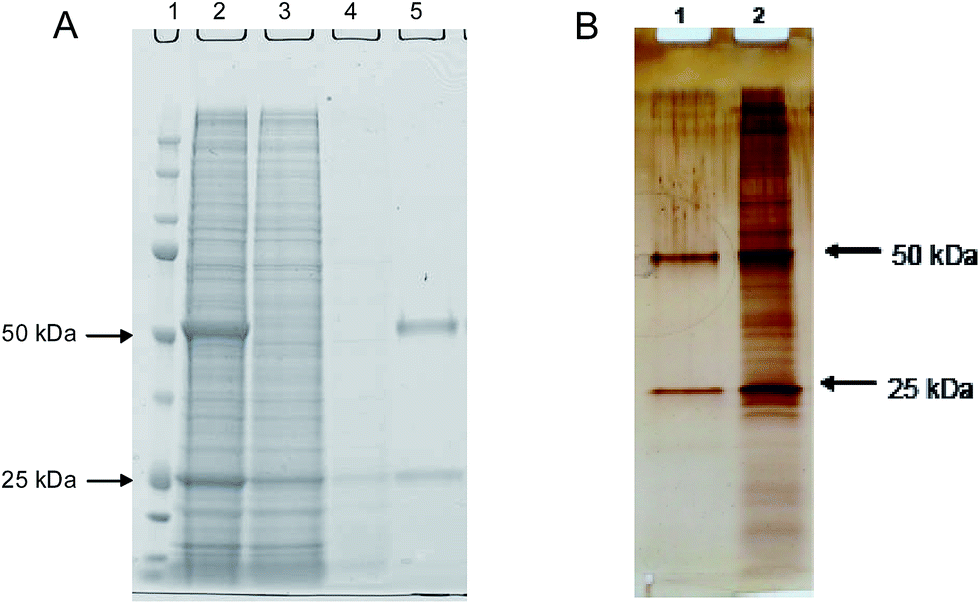 | ||
| Fig. 6 Results for SDS PAGE electrophoresis under reducing conditions of fractions obtained from (A) protein A affinity capture of IgG from cell culture supernatant. Lane 1: protein marker; Lane 2: cell culture supernatant; Lane 3: flow through fraction; Lane 4: wash fraction; Lane 5: elution fraction. (B) Imprinted membrane purification of IgG from cell culture supernatant. Lane 1: elution fraction; Lane 2: cell culture supernatant. Both gels were silver stained. | ||
Antibody capture using an epitope-imprinted membrane adsorber
The epitope (T10) imprinted membrane was subjected to a similar performance test using the protein A capture as a benchmark. Fig. 7A shows the breakthrough curves for six stacked MIP membranes. The dynamic binding capacity found for the six stacked membranes was 30 μg antibody per cm2 membrane area and 3.7 mg ml−1 membrane volume, the latter corresponding to nearly half the capacity of a commercial protein A macroporous membrane (5–7.5 mg ml−1).18 Because the commercial membrane has a smaller average pore size (0.45 μm) compared to the base membrane used in this study (1.0 μm), this difference can be attributed to the difference in specific surface of the supports. Because the established grafting method can easily be applied to supports of varied pore structure, this parameter and hence protein binding capacity can be adapted to the intended application.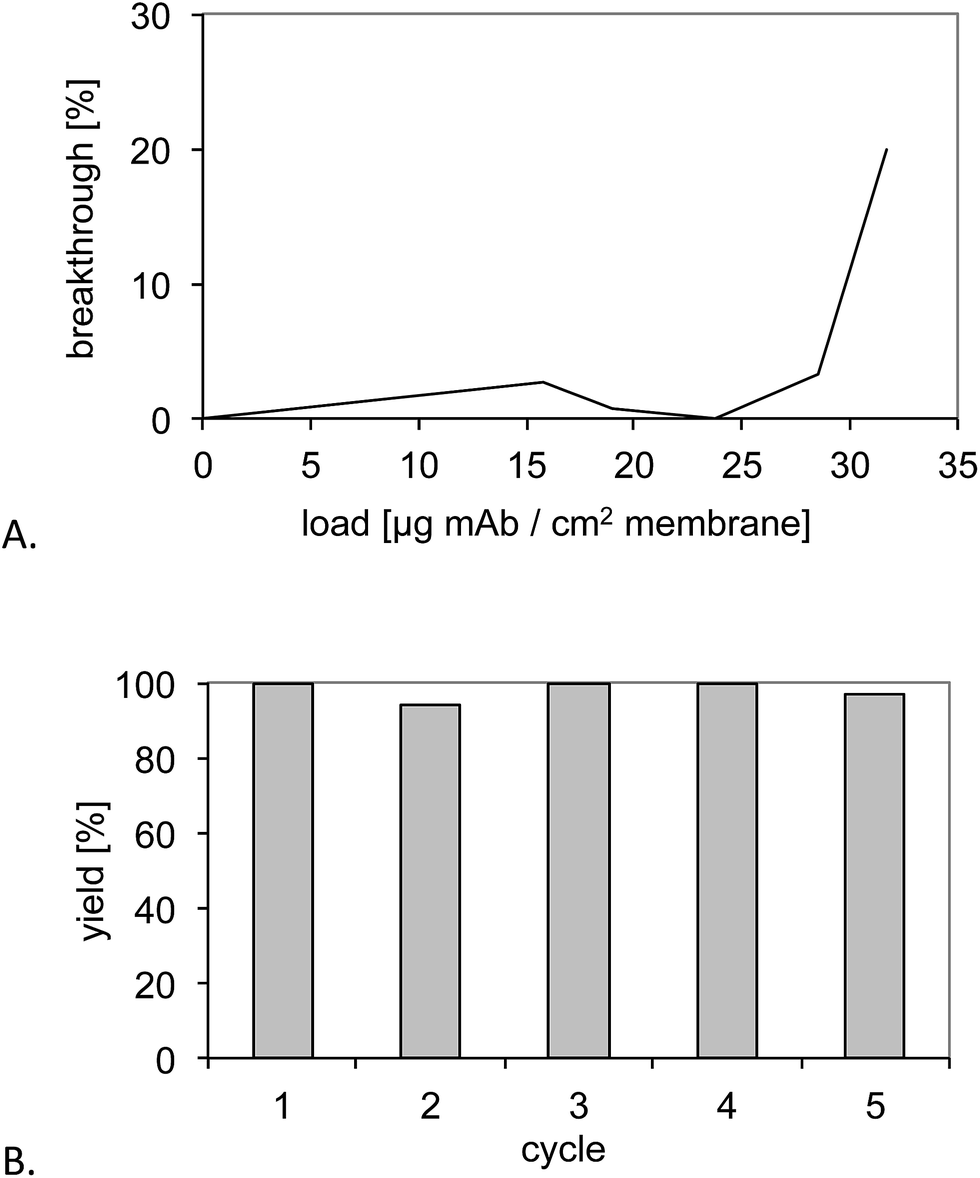 | ||
| Fig. 7 (A) Determination of the dynamic binding capacity of six stacked MIP membranes. (B) Recovery of the monoclonal antibody upon five purification cycles and intermediate cleaning with 0.5 M NaOH. | ||
Nevertheless, capacity was significantly lower compared to the conventional, “gold standard” protein A gel (cf. above). However this is typically observed for macroporous membranes vs. conventional chromatographic beads and can be related to their lower specific surface area. Macroporous membrane adsorbers have their main advantages in fast separations at lower buffer consumption.2,12,18
The membranes were then subjected to five purification cycles to test the stability towards conventional CIP procedures (Fig. 7B). This resulted in yields between 80% and 90% and no obvious deterioration of the performance.
To study the depletion of host cell proteins the antibody containing cell culture supernatant was loaded under low salt conditions. Using the imprinted membrane 88% (0.7–1.2 log units) impurity removal from the culture supernatant was achieved. The very obvious cleaning efficiency of the synthetic affinity membrane is also reflected in the SDS-PAGE electropherograms in Fig. 6B.
Conclusions
We have demonstrated a straightforward epitope imprinting procedure for the preparation of IgG affinity membranes directly applicable to mAb capture under near process realistic conditions. The procedure is based on the use of a C-terminal epitope of a conserved sequence of the constant region of IgG as template. Therefore we expect these membranes to display general affinity for monoclonal antibodies addressing the largest family of protein drugs today on the market and in the drug development pipeline. The performance of the membranes falls short of the protein A gold standard in terms of the slightly lower capacity and the lower efficiency of the host cell protein removal. In spite of these shortcomings the results are very promising and suggests strategies for further performance improvements. Hence we will further optimize monomer composition in order to enhance affinity and high salt compatibility. We expect this strategy to become widely applicable for tailormade down stream capture of biopharmaceuticals.Acknowledgements
This project was financially supported by the Bundesministerium für Bildung und Forschung (BMBF) under the project number: FKZ0315339A.Notes and references
- J. H. Chon and G. Zarbis-Papastoitsis, New Biotechnol., 2011, 28, 458–463 CrossRef CAS PubMed.
- P. Gronemeyer, R. Ditz and J. Strube, Bioengineering, 2014, 1, 188–212 CrossRef CAS.
- S. Ahuja, Handbook of bioseparations, Academic Press, Orlando, FA, 2000, vol. 2 Search PubMed.
- (a) G. El Khoury and C. R. Lowe, J. Mol. Recognit., 2013, 26, 190–200 CrossRef CAS PubMed; (b) A. C. A. Roque, M. Â. Taipa and C. R. Lowe, J. Chromatogr. A, 2005, 1064, 157–167 CrossRef CAS PubMed.
- Y. Liu, Y. Lu and Z. Liu, Chem. Sci., 2012, 3, 1467–1471 RSC.
- S. F. Teng, K. Sproule, A. Husain and C. R. Lowe, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 740, 1–15 CrossRef CAS.
- (a) N. W. Turner, C. W. Jeans, K. R. Brain, C. Allender, V. Hlady and D. W. Britt, Biotechnol. Prog., 2006, 22, 1474–1489 CrossRef CAS PubMed; (b) M. E. Byrne and V. Salian, Int. J. Pharm., 2008, 364, 188–212 CrossRef CAS PubMed; (c) D. E. Hansen, Biomaterials, 2007, 28, 4178–4191 CrossRef CAS PubMed; (d) T. Takeuchi and T. Hishiya, Org. Biomol. Chem., 2008, 6, 2459–2467 RSC; (e) Y. Ge and A. P. F. Turner, Trends Biotechnol., 2008, 26, 218–224 CrossRef CAS PubMed.
- (a) H. Nishino, C.-S. Huang and K. J. Shea, Angew. Chem., Int. Ed., 2006, 45, 2392–2396 CrossRef CAS PubMed; (b) Y. Li, H.-H. Yang, Q.-H. You, Z.-X. Zhuang and X.-R. Wang, Anal. Chem., 2006, 78, 317–320 CrossRef CAS PubMed; (c) H.-Y. Lin, C.-Y. Hsu, J. L. Thomas, S.-E. Wang, H.-C. Chen and T.-C. Chou, Biosens. Bioelectron., 2006, 22, 534–543 CrossRef CAS PubMed; (d) X. Hu, G. Li, J. Huang, D. Zhang and Y. Qiu, Adv. Mater., 2007, 19, 4327–4332 CrossRef CAS; (e) A. Nematollazadeh, W. Sun, C. S. A. Aureliano, D. Lütkemeyer, J. Stute, M. J. Abdekhodaie, A. Shojaei and B. Sellergren, Angew. Chem., Int. Ed., 2011, 50, 495–498 CrossRef PubMed; (f) D. Yin and M. Ulbricht, Biomacromolecules, 2013, 14, 4489–4496 CrossRef CAS PubMed; (g) R. Ouyang, J. Lei and H. Ju, Chem. Commun., 2008, 5761–5763 RSC; (h) Z. Yuan-Jin, Z. Xiang-Wei, H. Jing, L. Juan, X. Wen-Yu and G. Zhong-Ze, Angew. Chem., Int. Ed., 2009, 48, 7350–7352 CrossRef PubMed; (i) A. Cutivet, C. Schembri, J. Kovensky and K. Haupt, J. Am. Chem. Soc., 2009, 131, 14699–14702 CrossRef CAS PubMed.
- (a) A. Rachkov and N. Minoura, Biochim. Biophys. Acta, 2001, 1544, 255–266 CrossRef CAS PubMed; (b) M. M. Titirici, A. J. Hall and B. Sellergren, Chem. Mater., 2003, 15, 822–824 CrossRef CAS.
- S. Wei, et al., in preparation.
- H. Fröhlich, L. Villian, D. Melzner and J. Strube, Chem. Ing. Tech., 2012, 84, 905–917 Search PubMed.
- M. Ulbricht, Polymer, 2006, 47, 2217–2262 CrossRef CAS.
- Q. Yang, N. Adrus, F. Tomicki and M. Ulbricht, J. Mater. Chem., 2011, 21, 2783–2811 RSC.
- D. Yin and M. Ulbricht, J. Mater. Chem. B, 2013, 1, 3209–3219 RSC.
- S. Schwark and M. Ulbricht, Eur. Polym. J., 2012, 48, 1914–1922 CrossRef CAS.
- H. G. Heine and H. Rudolph, Liebigs Ann. Chem., 1971, 754, 28–34 CrossRef CAS.
- (a) M. R. Halhalli, C. S. A. Aureliano, E. Schillinger, C. Sulitzky, M. M. Titirici and B. Sellergren, Polym. Chem., 2012, 3, 1033–1042 RSC; (b) G. Wu, J. Li, X. Qu, Y. Zhang, H. Hong and C. Liu, RSC Adv., 2015, 5, 47010–47021 RSC.
- https://www.sartorius.de/de/product/product-detail/93prap06hb-12-a/ .
| This journal is © The Royal Society of Chemistry 2016 |