DOI:
10.1039/C6RA06616J
(Paper)
RSC Adv., 2016,
6, 66978-66989
Funnel shaped molecules containing benzo/pyrido[1,2,5]thiadiazole functionalities as peripheral acceptors for organic photovoltaic applications†
Received
12th March 2016
, Accepted 3rd July 2016
First published on 6th July 2016
Abstract
A series of four novel funnel shaped triphenylamine core derivatized materials with various combinations of 3,6-di-tert-butyl-9H-carbazole as donors and benzo/pyrido[1,2,5]thiadiazole as peripheral acceptors were designed, synthesized and characterized. Optical and electrochemical studies revealed that increasing acceptor strength in the structure leads to a bathochromic shift in their absorption profile and stabilizes the lowest unoccupied molecular orbital (LUMO) energy level. Enhancement in the intensity of an intramolecular charge transfer transition with synergistic downshift of the highest occupied molecular orbital (HOMO) and LUMO levels was observed by increasing the number of acceptor units. Thermogravimetric analyses (TGA) indicated that the materials containing pyrido[1,2,5]thiadiazole (PTD) acceptor functionality have excellent thermal stability compared to the materials containing benzo[1,2,5]thiadiazole (BTD) functionality. The measurement of glass transition temperature (Tg) corroborated TGA results and the values were found to be in the range of 148–198 °C. X-ray analyses indicated that more twist in the structure leads to absorption in the high energy region and varying the number of electron donor and acceptor groups has an impact on the modulation of frontier energy levels. A power conversion efficiency of 2.21% for the molecule with PTD as an acceptor and bis-carbazole as donor was achieved for the preliminary photovoltaic devices under simulated AM 1.5 illumination (100 mW cm−2).
Introduction
The current energy and environmental crisis due to the predicted exhaustion of fossil fuels has drawn world-wide attention towards clean, inexhaustible and long term securable renewable energy sources. Among several renewable energy technologies, solution-processable bulk-heterojunction (BHJ) devices and dye-sensitized solar cells have attracted considerable attention and are the most studied strategies.1,2 BHJ devices in particular are esteemed due to their advantages of being lightweight, having a low cost, and having flexibility in large-area applications.3,4 From the materials perspective, two types of π-conjugated organic materials, namely polymeric and small molecular entities, have been explored in developing the BHJ architecture. Polymers can exert extended photo-responses and good film forming capabilities with the conventional acceptors such as solvent-dissolvable fullerene derivatives, [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) and its C71 analogue (PC71BM). On the other hand polymeric donor materials pose number of challenges such as difficulty in purification, end group contamination and broad molecular weight distribution. Regardless, power conversion efficiency (PCE) values over 10% have been reported with such polymeric donors.5 However, in the interim, solution-processed small molecular donor-based BHJ devices have also aroused great interest, mainly due to their advantages of well-defined chemical structure, convenient purification methods and monodisperse molecular weight.6–9 Such advantages allow and encourage researchers to exert efforts to design and develop small molecular donors. Recently, Heliatek, a German company, announced an unprecedented cell efficiency of 12% using tandem solar cells featuring small molecules.10 This implies that small molecules may surmount their corresponding polymeric counterparts in the near future.
To improve the photovoltaic performance of BHJ devices, new synthetic strategies with rational molecular design and device optimization have been demonstrated.6,11,12 Among the several approaches emerged in molecular design, reducing the optical band gap for robust spectral coverage and tuning HOMO and LUMO levels of chromophores to achieve better energetic alignment with electron acceptors are paramount.13,14 Over the last decade, large number of small molecular electron donors have been extensively investigated with various molecular architectures such as donor–acceptor–donor (D–A–D),15–17 A–D–A,18–23 D–A–A,24–27 star shaped28–32 and X-shaped.33 We found that the star shaped molecules offer potential advantages like better miscibility with electron acceptors and well defined molecular packing with lesser extent of π–π aggregation due to their non-planar structure. However, these materials have received less attention due to their moderate photovoltaic performance in comparison with linear/branched molecules. PCE of 4.71% has been reported, which is the highest efficiency so far for the solution-processable BHJ devices using star shaped molecules.30 This implies that further scrutinization based on this module is needed. It is known that benzo/pyrido[1,2,5]thiadiazole groups have been used as central acceptor units in the materials for various organic electronic applications such as organic field effect transistors (OFET),34,35 dye-sensitized solar cells (DSSC)36 and BHJ solar cells.21,37,38 These acceptor units are known to increase electron affinity along with strong intermolecular π–π interactions which consequences in enhancing photovoltaic performance.39 Particularly for BHJSC applications, incorporation of benzo/pyrido[1,2,5]thiadiazole groups as peripheral units into a star shaped π-conjugated system is less explored to date. It is expected that incorporation of such acceptor functionalities can help target chromophore to acquire merits, such as good miscibility with fullerene acceptors and better interaction at D–A interface to obtain optimal charge transport in the desired direction.
Keeping these points in mind, we envisaged to design molecules with desymmetrization approach on the star shaped architecture by introducing various acceptors and π-building blocks. Herein, we report the design and development of a novel, funnel-shaped molecular framework that contains triphenylamine as the central core unit, which has been derivatized with 3,6-di tert-butyl-9H-carbazole (donor part) and benzo/pyrido[1,2,5]thiadiazole (acceptor part) functionalities (Fig. 1). The selection of tert-butyl group on carbazole was made to ensure sufficient solubility and to prevent π–π aggregation of the resultant chromophores.40 We have demonstrated the effect of pyrido[1,2,5]thiadiazole (PTD) or benzo[1,2,5]thiadiazole (BTD) units and their influence on optoelectronic properties. This study allows more comparative measures in evaluating structure–property relationship.
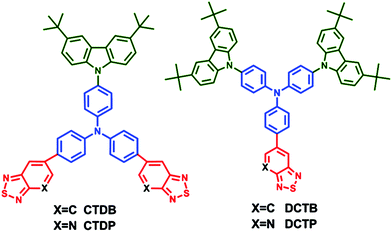 |
| | Fig. 1 Molecular structures of the newly designed and investigated materials. | |
Results and discussion
Design and synthesis
All of the materials were synthesized according to a multistep synthetic pathway depicted in Scheme 1. Synthesis of 5-bromobenzo[c][1,2,5]thiadiazole (1a) and 6-bromo[1,2,5]thiadiazole[3,4-b]pyridine (1b) was carried out by the ring closure reaction of SOCl2 with 4-bromobenzene-1,2-diamine and 5-bromopyridine-2,3-diamine, respectively, dissolved in pyridine at 0 °C.34 These bromo derivatives, 1a and 1b, were treated with n-butyl lithium at −78 °C to eliminate halogen atoms followed by the addition of 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxa-borolane in order to afford the corresponding boronate intermediates 2a and 2b. 3,6-Di-tert-butyl-9H-carbazole (3) was prepared according to the reported literature.41 Carbazole substituted dibromo derivative of triphenylamine (5) was synthesized using the classic Ullmann coupling reaction conditions.42 Syntheses of bromo derivatives 6a and 6b were carried out by the palladium catalyzed Suzuki coupling of boronate derivatives 2a and 2b with tris(4-bromophenyl)amine.28 Finally, the target dyes DCTB and DCTP were synthesized in excellent yields by stoichiometrical addition of 3 with 6a and 6b, respectively, using CuI/1,10-phenonthroline in the presence of K2CO3. The dyes CTDB and CTDP were obtained through the well-known palladium catalyzed Suzuki coupling of 5 with two equivalents of boronate derivatives 2a and 2b respectively.43 All these dyes were characterized by spectroscopic means using 1H NMR, 13C NMR, IR, EI-MS and MALDI-TOF. The details of the experimental and characterization data is available in the ESI.†
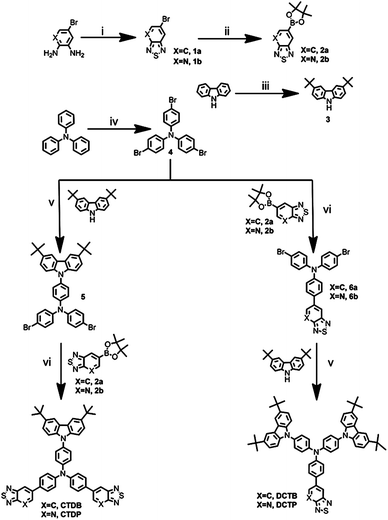 |
| | Scheme 1 Synthetic strategy for the preparation of target chromophores. Reagents and conditions: (i) SOCl2, pyridine, 0 °C; (ii) 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, n-BuLi, THF, −78 °C; (iii) t-BuCl, anhyd. AlCl3, CH3NO2, 24 h, rt; (iv) N-bromosuccinimide, DMF, 24 h, rt; (v) CuI, Cs2CO3, 1,10-phenonthroline, DMF, reflux, 24 h; (vi) Pd(PPh3)4, PhCH3/2 M K2CO3, 80 °C. | |
Molecular geometry and crystal structure
To further understand the structure–property relationship at molecular level, all the molecules were optimized using the B3LYP functional and 6-311G (d, p) basis set implemented in the Gaussian 09 program. From the optimized structures (Fig. 2b) it was observed that the average value of CNC angle was ∼120° in all the dyes, whereas mean value of dihedral angle between the planes of the central NCCC atoms was found to be more deviated with the torsion angle value of ∼45°. The carbazole unit attached to the TPA core was twisted in an out-of-plane fashion with an angle of ∼53° which is in accordance with the X-ray structure of CTDP (Fig. 2a). Comparative dihedral angles were obtained for the acceptor units attached to the TPA core and the values are measured to be ∼38° for the DFT optimized geometry and ∼34° for the crystal structure (CTDP). The optimized geometry of four molecules revealed that significant deviation from co-planarity between donor and acceptor fragments leads to break in conjugation which consequences the absorption at low wavelength region.44 The bond distances between C–N and C–C bonds in acceptor functionalities were computed to be 1.306 and 1.365 Å respectively, as predicted by the B3LYP level of theory. The variation with respect to acceptor groups is brought out clearly with shortening of this bond when an electron withdrawing group is introduced. The C–N bond is almost 0.059 Å shorter than C–C bond and the values are given in Fig. S1 (ESI†). The single X-ray crystal structure of the intermediate 6b is given Fig. S2 (ESI†). The longer wavelength absorption is originated mainly from HOMO−1 and HOMO levels to LUMO level in case of all the dyes. The molecular orbital amplitude plots of FMOs such as HOMO−1, HOMO and LUMO, are shown in Fig. 3. The electron density of HOMO−1 in DCTB and DCTP is predominantly populated on carbazole unit. On the other hand in dyes CTDB and CTDP, it is mainly delocalized over the carbazole and TPA units. In HOMO of all the dyes, electron density is delocalized over the carbazole and TPA groups, whereas LUMO is predominantly localized on acceptor units. The presence of well separated electron density from HOMO to LUMO is a strong indication of better intramolecular charge transfer transition. The larger transition dipole moment of the dyes indicates that these molecules are more polarized in excited state which is in accordance with our emission data (positive solvatochromism).
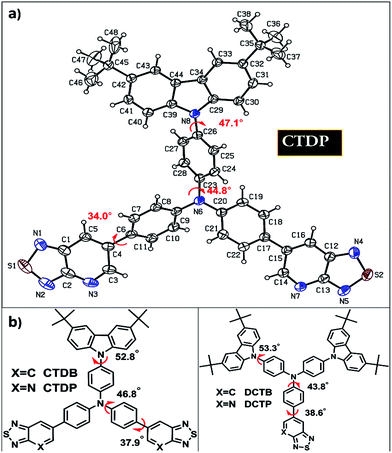 |
| | Fig. 2 (a) ORTEP diagram of CTDP with the atom-numbering scheme and dihedral angle values. Displacement ellipsoids are drawn at the 30% probability level for non-hydrogen atoms, and H atoms are shown as small spheres of arbitrary radius; (b) DFT optimized geometries from B3LYP/6-311g (d, p) level of theory. | |
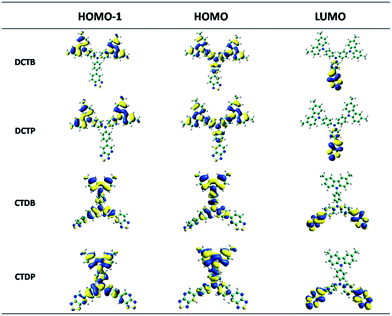 |
| | Fig. 3 Computed isodensity (0.02) surfaces of the HOMO−1, HOMO and LUMO of the target molecules. | |
Photophysical properties
The absorption and emission spectra of these four dyes recorded in toluene are shown in Fig. 4 and the salient features of the corresponding spectra are summarized in Tables 1 and S1 (ESI†). The absorption spectra of these dyes showed two distinct bands. The first absorption peak at 312–323 nm is attributed to π → π* transition of the conjugated backbone. The second absorption peak of the dyes DCTB and CTDB containing BTD unit displayed maximum wavelength (λmax) at 410 nm and the dyes DCTP and CTDP containing PTD acceptor unit showed λmax at 435 nm which can be assigned to ICT transition from donor (carbazole) to acceptor part.45–47 The bathochromic shift of 25 nm in the λmax of DCTP and CTDP dyes can be attributed to the presence of electron withdrawing pyridine in the PTD unit. Moreover, increased number of acceptor groups in the studied structures has pronounced effect in the ICT peak intensity i.e., molar extinction coefficients were increased to two-fold (Fig. 4). It has been noticed that absorption spectra remain unchanged irrespective of any variations in donor/acceptor units of the target dyes. As expected, going from solution to film absorption, the absorption spectra of all the dyes were red shifted (Fig. S3, ESI†) which indicate the presence of strong intermolecular packing in the solid state. The optical band gap (Eg) values are calculated using the onsets of thin film absorption spectra.
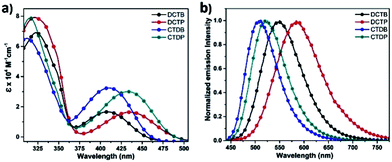 |
| | Fig. 4 (a) Absorption and (b) emission spectra of dyes (1 × 10−5 M) recorded in toluene at ambient temperature. | |
Table 1 Photophysical properties of the dyes
| Dye |
λmaxa (nm) |
ε (M−1 cm−1) |
λfilma (nm) |
Eg,optb (eV) |
| Absorption spectra of the dyes (1 × 10−5 M) were recorded in toluene at ambient temperature. Estimated from the onset absorption spectra of the dyes in thin film state. |
| DCTB |
409 |
16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 639 639 |
434 |
2.43 |
| 323 |
69138 |
| DCTP |
435 |
16456 |
450 |
2.30 |
| 321 |
79147 |
| CTDB |
410 |
32459 |
434 |
2.49 |
| 312 |
65299 |
| CTDP |
433 |
30022 |
442 |
2.27 |
| 316 |
78770 |
To examine the electronic and spectroscopic properties, optimized geometries were used for the computation of vertical excitation energies using TDDFT with various functionals such as B3LYP, PBE, M06-2X and 6-311g (d, p) basis set.48 Among them, M06-2X in the framework of PCM using toluene as solvent showed reasonable agreement with small underestimation and the trend is consistent with experimental observation, whereas B3LYP and PBE are highly over-estimated.49 The computed energies of vertical excitations along with oscillator strength and compositions in terms of molecular orbitals are compiled in Table 2. The comparison of normalized plots of simulated spectra with oscillation strength and experimental absorption of dyes are depicted in Fig. S4 ESI.† The oscillation strength of these dyes gradually increased by increasing the number of acceptor groups in the molecular backbone which corroborates our experimental absorption.
Table 2 Theoretical data of the dyes
| Dye |
λmaxa (nm) |
M06-2X (toluene) |
Composition |
μgb (Debye) |
μec (Debye) |
| λmax (nm) |
f |
| Absorption values measured in toluene. Ground state dipole moment of the dyes obtained from B3LYP functional. Transition dipole moment of the dyes. |
| DCTB |
409 |
370 (3.35 eV) |
0.59 |
H → L (67%) |
1.61 |
1.78 |
| 323 |
H−1 → L (17%) |
| DCTP |
435 |
392 (3.17 eV) |
0.52 |
H → L (76%) |
3.25 |
3.68 |
| 321 |
H−1 → L (17%) |
| CTDB |
410 |
375 (3.31 eV) |
0.66 |
H → L (67%) |
2.52 |
2.85 |
| 312 |
H−1 → L (14%) |
| CTDP |
433 |
394 (3.15 eV) |
0.69 |
H → L (67%) |
1.31 |
1.49 |
| 316 |
H−1 → L (19%) |
To study the effect of solvatochromism, absorption and emission spectra of all the dyes were recorded in a variety of solvents with different empirical solvent polarity parameter [ET(30)] such as hexane, toluene, chloroform, THF, acetonitrile, DMF and methanol (Fig. S3 and S5, ESI†) and the corresponding data is provided in Tables S2–S4, ESI.† The absorption spectra of all the dyes are impassive irrespective of solvent polarity. In emission spectra, significant positive solvatochromism is observed for these dyes, i.e., emission maximum becomes broad and red shifted as the polarity of solvent increases from non-polar to polar aprotic solvent, implying strong ICT transition between donor and acceptor counterparts. Non-polar locally excited (LE) state, as seen in absorption, is transformed into a polar excited state, as evident from emission, in the presence of apolar aprotic solvent. On the other hand, the photoluminescence was completely quenched with polar protic solvents like DMF and methanol (Tables S3 and S4, ESI†). This may be attributed to the interaction of polar protic solvent dipole with extensive polarization induced by thiadiazole acceptor unit which lead to non-radiative relaxation.50
Fluorescence lifetime is that characteristic time in which a molecule can undergo conformational changes as well as interaction with other molecules or diffuse through the local environment. The performance of organic solar cells is greatly influenced by the dynamics of excitons which is pertinent to the fluorescence lifetime of the dyes. The excited-state lifetimes of all the four dyes were measured in toluene (Fig. 5a) and the values were found to be 6.61, 7.29, 5.11 and 7.03 ns for DCTB, DCTP, CTDB and CTDP respectively. The gradual increment in emissive lifetime was observed by replacing benzene with pyridine in the acceptor unit. The electron-transfer rates (kET) of these molecules were found in the range of 1.4–2.0 × 108 s−1 (Table S1 ESI†).
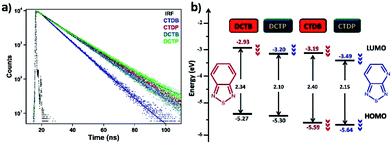 |
| | Fig. 5 (a) Time-resolved fluorescence decay curves of the dyes in toluene (λex = 405 nm, λem = 530 nm); (b) schematic diagram represents the modulation of energy levels estimated from electrochemical data. | |
Electrochemical properties
The redox behaviour of target dyes is essential to investigate the alignment of these frontier energy levels with respect to the energy levels of electron acceptors, such as PC61BM, for efficient charge transport. Cyclic voltammetry was performed to evaluate oxidation and reduction potentials using three-electrode configuration. Fig. 6 shows the resulting cyclic voltammogram (CV) traces. The CVs of dyes DCTB and DCTP showed three quasi-reversible oxidations which are originated from two carbazole donor and triphenylamine units. Whereas dyes CTDB and CTDP showed two quasi-reversible oxidations and the oxidation peaks are attributed to carbazole donor and triphenylamine unit. All the dyes showed single reduction potential. Electrochemical band gaps were estimated from the peak potentials of anodic and cathodic waves based on −4.4 eV as SCE energy level relative to vacuum. The first oxidation and reduction peak potentials were used to calculate the HOMO and LUMO energy levels. The CV data of DCTB–DCTP and CTDB–CTDP transformations was compared to examine that how the variation in number of units and acceptor strength involves in modulation of frontier energy levels. For instance, in DCTB–DCTP and CTDB–CTDP transformations, the replacement of BTD by PTD unit induced the anodic peak potential (Epa) with a positive shift of 30–40 mV and 290 mV in cathodic peak potential (Epc). This indicated that introduction of PTD unit has minute influence on HOMO level, albeit affects the LUMO level greatly. For DCTB–CTDB transformation, a shift is observed in anodic peak (Epa) from 0.87 to 1.19 V and same pattern is observed in DCTP–CTDP transformation i.e., anodic peak (Epa) was shifted from 0.9 to 1.23 V which indicates the positive shift of 320–330 mV upon increasing the number of acceptor units. The potential of cathodic peak (Epc) shifted toward more positive values from −1.47 to −1.20 V for DCTB–CTDB transformation and −1.18 to −0.91 V for DCTP–CTDP transformation corresponding to the value of ∼270 mV. Similar trend was observed in triphenylamine–thienylvinylene hybrid compounds reported by Roncali et al.29,51–54 The HOMO and LUMO energy levels of these materials were calculated from the onsets of their corresponding oxidation and reduction potentials. The results of CV analyses revealed that these dyes display oxidation and reduction at more positive potentials with increasing the number of acceptor units and induce a simultaneous downshift of both HOMO and LUMO levels. Observed HOMO and LUMO energy levels are well matched with the commonly used acceptor material PCBM. Since the LUMO levels of the newly synthesized materials are deeper compared to the threshold value of 0.3 eV required for effective charge separation, efficient electron transfer is expected from these materials to PCBM and is beneficial to improve short-circuit current (Jsc) and open circuit voltage (Voc).21,55 The electrochemical band gaps observed for DCTB and DCTP were comparatively low when compared with CTDB and CTDP, which indicates that band gap (ΔE) increases with number of acceptor units and decreases with acceptor strength (Fig. 5b). It is worth noting that the electrochemical and optical band gap values are relatively in good agreement. Theoretically predicted LUMO energy levels are about 0.3 eV low, whereas the HOMO energy levels match well with the electrochemical data and the values are summarized in Tables 3 and S5, ESI.†
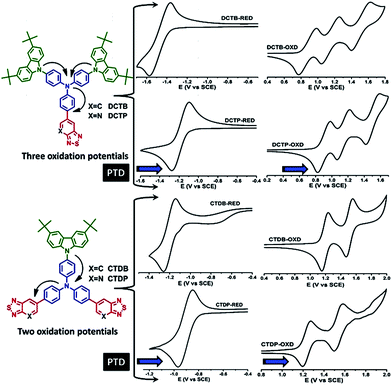 |
| | Fig. 6 Cyclic voltammograms of the dyes recorded in dichloromethane. | |
Table 3 Electrochemical data of the dyes
| Dye |
Eoxd1/2a (V) |
Ered1/2a (V) |
HOMOb (eV) |
LUMOb (eV) |
Eg,elec (eV) |
| 1st |
2nd |
3rd |
| Measured in CH2Cl2 with 0.1 M tetrabutylammonium perchlorate (TBAPC) as supporting electrolyte with a scan rate between 50 mV s−1. Deduced from the formula HOMO = −(4.4 + Eox) and LUMO = −(4.4 + Ered). Bandgap obtained from electrochemical data. |
| DCTB |
0.87 |
1.15 |
1.28 |
−1.47 |
−5.27 |
−2.93 |
2.34 |
| DCTP |
0.90 |
1.14 |
1.51 |
−1.18 |
−5.30 |
−3.20 |
2.10 |
| CTDB |
1.19 |
1.51 |
— |
−1.20 |
−5.59 |
−3.19 |
2.40 |
| CTDP |
1.23 |
1.54 |
— |
−0.91 |
−5.64 |
−3.49 |
2.15 |
Thermal properties
The thermal properties and phase transition nature of all the dyes were evaluated by thermogravimetric analysis and differential scanning calorimetry, respectively, under nitrogen atmosphere at a heating rate of 10 °C min−1. All the dyes exhibited excellent thermal stability. The temperature corresponding to 5% weight loss for the dyes is in the range of 389–515 °C.56,57 The dyes having PTD unit undergo less thermal degradation and possess higher decomposition temperatures (Fig. 7) and the data is provided in Table S6, ESI.† The high thermal stability of DCTP and CTDP may be attributed to the well-ordered π–π interaction between the neighbouring units. In other words, the possibility of interaction sites in PTD are pyridine and thiadiazole units which lead to efficient intermolecular interactions. On the other hand, these intermolecular interactions are expected to be weak in the case of the dyes containing BTD units. The observation identified in TGA curves of DCTB and CTDB (Fig. 7a) may due to solvent/moisture associated with the chemical, is being ejected. Melting point (Tm) values were found to be in the range of 313–371 °C. The glass-transition temperature (Tg) values were observed in the range of 148–198 °C which is determined from the second heating scan and gradually increased in the order of CTDB < CTDP < DCTB < DCTP.58 This is attributed to the presence of more tertiary substitution on carbazole unit which increases the amorphous nature of the local system.59
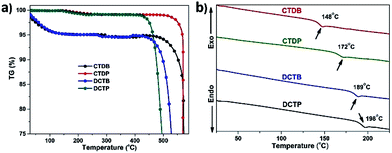 |
| | Fig. 7 Thermograms (a) and DSC traces (b) of the dyes measured at a heating rate of 10 °C min−1 under N2 atmosphere. | |
Photovoltaic properties
Because of their appropriate optoelectronic features, adequate solubility and good film-forming capabilities, the materials reported herein were incorporated as p-type semiconducting components along with soluble fullerene derivative [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) as a n-type semiconductor in BHJ photovoltaic devices. BHJ architectures typically deliver higher device power conversion efficiencies by maximizing the surface area of the interface between the donor and acceptor materials in the active layer. For all the compounds, the device structure used was ITO/PEDOT:PSS (38 nm)/active layer/Ca (20 nm)/Al (100 nm), where the active layer was a solution-processed blend of either of the donor materials and the solubilised fullerene PC61BM. The active layers were prepared by spin coating the mixtures of appropriate material with PC61BM in chlorobenzene under ambient conditions @ 1:1 ratios without subsequent annealing. The optimum layer thickness was found to be in the range of approximately 60 nm. We chose a high boiling solvent for casting an active layer on top of the PEDOT:PSS surface as our attempts to fabricate devices using low boiling solvents, such as CHCl3, resulted in very poor photovoltaic performance. This was primarily due to low film quality. Moreover, the use of high-boiling solvent is preferable from a processing point of view. Though we fabricated our devices using all of the materials reported in this study, we were not able to fabricate CTDB using chlorobenzene, primarily due to inadequate solubility. CTDB showed enough solubility in CHCl3, however the film interpretation was inconsistent to fabricate any devices. All other materials, CTDP, DCTB and DCTP, showed miscibility with PC61BM in their blend solutions, hence used to construct BHJ devices.
The best photovoltaic devices based on DCTP, where PTD acceptor was used, gave power conversion efficiency (PCE) of 2.21% whereas its analogue DCTB afforded 1.06%. The BHJ devices based on DCTP and DCTB showed similar open circuit voltages (Voc) which is an observation consistent with the measured HOMO values. Devices based on DCTP showed higher photocurrent than the devices based on DCTB, a finding that is consistent with the observed bathochromic-shift in the absorption spectrum of DCTP when compared with DCTB.
Overall comparison of the BHJ performance of DCTP and DCTB indicated that the use of PTD accepting unit can indeed provide a platform for achieving higher PCE and Jsc, when studied for a given donor–acceptor module. Our results clearly advocate the promotion of PTD as an interesting structural concept for the design and development of efficient BHJ materials. By contrast, the maximum PCE obtained for a device based on CTDP was 1.50%, when fabricated under similar conditions. It is notable to mention that the device based on CTDP afforded higher photocurrent than the device based on DCTB, an apparent observation corroborating the absorption profile of CTDP when compared with DCTB. The relatively lower efficiency achieved with the use of BTD acceptor when compared with PTD acceptor is likely due to the weaker absorption of the visible spectrum. This suggests that the light harvesting ability of small molecular donors plays a significant role in achieving their optimum photovoltaic performance. The respective current–voltage (J–V) curves are shown in Fig. 8, and Table 4 represents a comparative photovoltaic performance data.
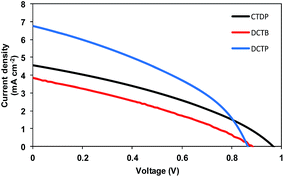 |
| | Fig. 8 Characteristic current density versus voltage (J–V) curves for the best BHJ devices based on CTDP (black), DCTB (red) and DCTP (blue) in blends with PC61BM under simulated sunlight (100 mW cm−2, AM 1.5G). The device structure is: ITO/PEDOT:PSS (38 nm)/active layer/Ca (20 nm)/Al (100 nm). | |
Table 4 Comparative BHJ solar cell performance of the dyes
| Dye |
Blend film thicknessa (1:1 @ 3000 rpm) |
Voc (mV) |
Jsc (mA cm−2) |
FF |
PCE% |
| BHJ devices with specified weight ratio. Device structure is ITO/PEDOT:PSS (38 nm)/active layer/Ca (20 nm)/Al (100 nm) with active layer thickness of about 65 nm. |
| CTDP |
63 |
970 |
4.55 |
0.34 |
1.50 |
| DCTB |
65 |
870 |
3.80 |
0.32 |
1.06 |
| DCTP |
64 |
871 |
6.75 |
0.38 |
2.21 |
Conclusions
In summary, we have developed a series of four molecules with funnel architecture which consists of triphenylamine as the central unit with various combinations of donor (3,6-di-tert-butyl-9H-carbazole) and acceptor (benzo/pyrido[1,2,5]thiadiazole) fragments. These materials were synthesized by classic Ullmann and palladium-catalysed Suzuki coupling reactions with good yields and were thoroughly characterized. The optical and electrochemical properties of the dyes are well tuned based on the strength and number of donor and acceptor units. The fluorescence quantum yields and excited state life times, and their electron-transfer rate constants were calculated to evaluate their excited state properties. The band gap obtained from cyclic voltammetry analysis is consistent with optical and theoretical data. The structure–property relationship of the dyes was investigated in detail using the results obtained from the experimental and DFT calculations. When these new materials were tested in solution-processable BHJ devices, better performance was observed with materials comprising PTD unit and these results corroborate the experimental and theoretical findings. The major highlight of this preliminary study revealed that nonetheless dyes are absorbing at 435 nm with low molar absorptivity, it reached the efficiency of 2.21%. Efforts are ongoing to further improve the PCE of devices and future studies will look at extending the conjugation in the studied architecture towards longer wavelength region.
Experimental section
Materials and general procedures
Unless otherwise specified, reactions were performed under nitrogen atmosphere with standard Schlenk techniques. All the reagents were of reagent/analytical grade and used without further purification. Tetrahydrofuran (THF) and toluene were distilled over sodium and benzophenone, and N,N′-dimethyl-formamide (DMF) was dried over 3 Å molecular sieves under nitrogen atmosphere. All chromatographic separations were carried out on silica gel (60–120 mesh). 1H and 13C NMR spectra were measured on a Bruker Advance DPX spectrometer using CDCl3 solvent with TMS as internal standard. Mass spectra were obtained using electron ionization (EI) mass spectrometry (Thermofinnigan, Sanzox, CA), gas chromatography-mass spectrometry (GC-MS) was performed on VG70-70H. MALDI-TOF mass spectra were obtained from Axima performance MALDI-TOF/TOF mass spectrometer (Shimadzu). UV-Vis absorption spectra were measured on a Perkin-Elmer spectrophotometer and fluorescence spectra were recorded using a Spex model Fluorolog-3 spectrofluorometer. Fluorescence quantum yields were estimated by integrating the fluorescence bands and using 9,10-diphenyl anthracene (ϕ = 0.82 in toluene) as reference. Fluorescence lifetime measurements were performed on a picosecond time-correlated single photon counting (TCSPC) setup (Edinburgh Instruments Ltd, Lifespec II model) employing a picosecond light emitting diode lasers (Nano LED, λex = 405 nm) as the excitation source. The decay curves were recorded by monitoring the fluorescence emission maxima of the dyes. The lamp profile was recorded by placing a scatter (diluted Ludox solution in water) in place of the sample. The width of the instrument response function (IRF) was limited by the full width at half maxima (FWHM) of the excitation source, 625 ps at 405 nm. The quality of the fits was judged by χ2 values and the distribution of residuals. Perkin-Elmer Spectrum BX spectrophotometer was used to obtain IR spectra of the dyes at a resolution of 4 cm−1. Thermogravimetric analyses (TGA) were performed with a TGA/SDTA 851e (Mettler Toledo) thermal analyzer with a heating rate of 10 °C min−1 under nitrogen atmosphere in the temperature range of 33–550 °C. The glass-transition temperatures (Tg) were measured at a heating rate of 10 °C min−1 under nitrogen atmosphere using differential scanning calorimetry (DSC Q200 TA). Tg was determined from the second heating scan. Melting points were measured with an Electro thermal IA 9100 series digital melting point instrument and are uncorrected. Cyclic voltammetry measurements were performed on a PC-controlled CH instruments model CHI 620C electrochemical analyzer using 1 mM dye solution in dichloromethane at a scan rate of 100 mV s−1 using 0.1 M tetrabutylammonium perchlorate (TBAP) as supporting electrolyte. The glassy carbon, standard calomel electrode (SCE) and platinum wire were used as working, reference and auxiliary electrodes respectively.
X-ray crystallographic details
X-ray data for two compounds was collected at room temperature using a Bruker Smart Apex CCD diffractometer with graphite monochromated MoKα radiation (λ = 0.71073 Å) with ω-scan method. Preliminary lattice parameters and orientation matrices were obtained from four sets of frames. Unit cell dimensions were determined using 6154 reflections in DCTP and 5956 reflections in intermediate 6b crystal data. Integration and scaling of intensity data were accomplished using SAINT program. Structures were solved by Direct Methods using SHELXS97 and refinement was carried out by full-matrix least-squares technique using SHELXL97. Anisotropic displacement parameters were included for all non-hydrogen atoms. All H atoms were positioned geometrically and treated as riding on their parent C atoms with C–H distances of 0.93–0.97 Å, and with Uiso(H) = 1.2Ueq(C) or 1.5Ueq for methyl atoms. The crystallization solvent toluene was included in the crystal lattice of the compound. The solvent is found to be severely disordered to obtain a reliable structural model. The SQUEEZE utility in PLATON was used to remove contribution from the disordered solvent. An electron count of 110 was suggested per unit cell (void volume of 460 Å3) which closely relates to two molecules of toluene. The files (.hkl and .ins) generated by PLATON after the SQUEEZE treatment were used for the final refinement of structures.
Computational details
Density Functional Theory (DFT) calculations were performed using the Gaussian 09 (G09) ab initio quantum chemical software package. Initially, structural optimizations were performed without any symmetry constraints in gas phase. The local minima were further confirmed by vibrational analysis. For DFT calculations, Beckes three-parameter (B3) gradient-corrected exchange functional combined with Lee–Yang–Parr (LYP) correlation functional60,61 and 6-311G (d, p) basis set was used. Methyl groups were introduced instead of tert-butyl groups on carbazole to reduce computational cost. The minimized geometries were then used to obtain frontier molecular orbitals (FMOs) and also subjected to single-point time-dependent DFT (TDDFT) studies. The evaluation of 25 vertical singlet–singlet transitions was carried out to simulate the UV-Vis spectra of the dyes. The integral equation formalism polarizable continuum model (PCM) within the self-consistent field theory has been used to describe the solvation of dyes in toluene.62,63 The TDDFT calculations were performed with various energy functionals such as B3LYP, PBE and M06-2X.64 The simulation of the major portion of absorption spectrum and interpretation of the nature of transitions was executed using software GaussSum 2.2.5.65 The percentage contribution of individual fragments present in a given dye was calculated against their respective molecular orbitals.
Device fabrication and characterization of photovoltaic devices
Indium tin oxide (ITO)-coated glass (10 ohms per square) was cleaned by sonicating in a stirred solution of 5% (v/v) Deconex 12PA detergent for 30 min. The ITO-coated glass was then successively sonicated for 10 min each in distilled water, acetone, and isopropanol. The substrates were then cleaned by UV/ozone at room temperature for 10 min. The UV/ozone cleaning of glass substrates was performed using a Novascan PDS-UVT UV/ozone cleaner with the platform set to maximum height. The intensity of the lamp was >36 mW cm−2 at a distance of 10 cm. At ambient conditions, the ozone output of the UV cleaner is greater than 50 ppm. Aqueous solutions of PEDOT/PSS (HC Starck, Baytron P AI 4083) were filtered (0.2 μm RC filter) and deposited onto glass substrates in air by spin coating (Laurell WS-400B-6NPP lite single wafer spin processor) at 4000 rpm for 60 s to give a layer having a thickness of 40 ± 5 nm. The PEDOT/PSS layer was then annealed on a hotplate in a glove box at 120 °C for 10 min. For OPV devices, the newly synthesized organic p-type materials and PC61BM (Nano-C) were separately dissolved in individual vials by magnetic stirring. Blend ratios and solution concentrations were varied to optimize device performance. The solutions were then combined, filtered (0.2 μm RC filter), and deposited by spin coating (SCS G3P spin coater) onto the ITO-coated glass substrates inside a glove box (with H2O and O2 levels both <1 ppm). Film thicknesses were determined on identical samples using a Dektak 6M Profilometer. The coated substrates were then transferred (without exposure to air) to a vacuum evaporator inside an adjacent nitrogen-filled glove box. Samples were placed on a shadow mask in a tray. The area defined by the shadow mask gave device areas of exactly 0.1 cm2. Deposition rates and film thicknesses were monitored using a calibrated quartz thickness monitor inside the vacuum chamber. Aluminium (Al) (3 pellets of 99.999%, KJ Lesker), from an alumina-coated graphite boat, having thicknesses of 100 nm, respectively, were evaporated onto the active layer by thermal evaporation at pressures less than 2 × 10−6 mbar. Where used, C60 (Nano-C) and 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (Aldrich) were evaporated from alumina crucibles. A connection point for the ITO electrode was made by manually scratching off a small area of the active layers. A small amount of silver paint (Silver Print II, GC Electronics, part number: 22-023) was then deposited onto all of the connection points of both ITO and Al. The completed devices were then encapsulated with glass and a UV-cured epoxy (Summers Optical, Lens Bond type J-91) by exposure to 365 nm UV light inside the glove box for 10 min. The encapsulated devices were then removed from the glove box and tested in air within 1 h. Electrical connections were made using alligator clips. The OPV devices were tested using an Oriel solar simulator fitted with a 1000 W xenon lamp filtered to give an output of 100 mW cm−2 at simulated AM 1.5. The lamp was calibrated using a standard filtered silicon (Si) cell from Peccell Limited, which was subsequently cross-calibrated with a standard reference cell traceable to the National Renewable Energy Laboratory. The devices were tested using a Keithley 2400 source meter controlled by Labview software.
5-Bromobenzo[c][1,2,5]thiadiazole (1a)
Thionyl chloride (SOCl2) (2.91 mL, 37.4 mmol) was added drop wise to a stirred, cooled solution of 4-bromobenzene-1,2-diamine (5 g, 26.7 mmol) in pyridine (30 mL) at 0 °C and the reaction mixture was stirred for additional 2 h at this temperature. Then the reaction mixture was heated to reflux for 3 h and poured into water. The crude product was collected by filtration and washed with water thoroughly to remove excess of pyridine and SOCl2. The crude solid was purified by silica gel column chromatography using n-hexane as eluent affording 1a as a while solid (5.2 g, 91%). 1H NMR (500 MHz, CDCl3): δ 8.18 (s, 1H), 7.86–7.83 (d, 1H, J = 9.3 Hz), 7.65–7.60 (d, 1H, J = 9.3 Hz); 13C NMR (125 MHz, CDCl3): δ 155.2, 153.3, 133.1, 124.5, 123.8, 122.1; GC/MS: m/z 214 (M+).
6-Bromo-[1,2,5]thiadiazolo[3,4-b]pyridine (1b)
This compound was synthesized according to the procedure reported for 1a, using 5-bromopyridine-2,3-diamine (5.0 g, 26.5 mmol), to give the desired product as a light yellow solid (5.6 g, 98%). 1H NMR (500 MHz, CDCl3): δ 9.09 (s, 1H), 8.56 (s, 1H); 13C NMR (125 MHz, CDCl3): δ 159.9, 156.8, 147.8, 131.2, 121.1; GC/MS: m/z 215 (M+).
5-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[c][1,2,5]thiadiazole (2a)
n-Butyllithium (4.67 mL, 10.24 mmol (2.0 M solution in hexanes)) was added drop wise to a solution of 1a (2.0 g, 9.34 mmol) in anhydrous THF (25.0 mL) at −78 °C (dry ice-acetone bath) and the resultant yellow suspension was left to stir at this temperature for 2 h. 3.25 mL (4.19 mmol) of 2-isopropoxy-4,4,5,5-tetramethyl-[1,3,2]-dioxaborolane was added rapidly to this solution and the resulting reaction solution was stirred for 1 h at −78 °C followed by overnight stirring at room temperature. Saturated ammonium chloride (40 mL) and chloroform (50 mL) were added to the reaction mixture and the organic layer was separated. Organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, concentrated to a crude and subjected to chromatography on silica eluting with 5% ethyl acetate in hexanes to provide 2a as a white solid (1.9 g, 78%). 1H NMR (500 MHz, CDCl3): δ 8.50 (s, 1H), 7.97–7.91 (m, 3H), 1.39 (s, 12H); 13C NMR (125 MHz, CDCl3): δ 156.1, 133.6, 129.4, 120.6, 84.4, 24.9; GC/MS: m/z 262 (M+).
6-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-[1,2,5]thiadiazole[3,4-b]pyridine (2b)
This compound was synthesized according to the procedure reported for 2a, using 1b (2.0 g, 9.3 mmol), to give the desired product as a light yellow solid (2.1 g, 86%). 1H NMR (500 MHz, CDCl3): δ 9.36 (s, 1H), 8.78 (s, 1H), 1.48 (s, 12H); 13C NMR (125 MHz, CDCl3): δ 159.9, 147.1, 137.5, 83.4, 24.9; GC/MS: m/z 263 (M+).
3,6-Di-tert-butyl-9H-carbazole (3)
To a stirred solution of carbazole (10.0 g, 59.8 mmol) and anhydrous AlCl3 (24.4 g, 179.4 mmol) in nitromethane, tert-butyl chloride (19.7 mL, 179.4 mmol) was added under N2 atmosphere. The solution was stirred at room temperature for 18 h. The mixture was poured into water and extracted with chloroform. The combined organic layer was neutralized with sodium carbonate solution. Obtained organic solution was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The green residue obtained was coated with silica gel and purified by column chromatography using n-hexane to yield a white solid. The obtained product was further purified by recrystallization from CH2Cl2/hexane mixture to give white crystals (13.2 g, 47.3 mmol, 79%). 1H NMR (500 MHz, CDCl3): δ 8.07 (s, 2H), 7.66 (s, 1H, (N–H)), 7.45–7.43 (d, 2H, J = 8.5 Hz), 7.25–7.22 (d, 2H, J = 8.5 Hz), 1.42 (s, 18H). GC/MS: m/z 279 (M+).
Tris(4-bromophenyl)amine (4)
In a two-neck flask, NBS (23.9 g, 134.5 mmol) was added in portions to a solution of TPA (10.0 g, 40.8 mmol) in DMF (120 mL) at 0 °C. The mixture was warmed to room temperature, stirred for an additional 12 h and then poured into excess water. This aqueous solution was extracted with chloroform. The combined extracts were washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography using n-hexane as eluent to yield a white solid (17.8 g, 37.0 mmol, 91%). 1H NMR (500 MHz, CDCl3): δ 7.36–7.33 (dd, 6H, J = 8.9 Hz), 6.93–6.90 (dd, 6H, J = 8.9 Hz). GC/MS: m/z 481 (M+).
4-Bromo-N-(4-bromophenyl)-N-(4-(3,6-di-tert-butyl-9H-carbzol-9-yl)phenyl)aniline (5)
A mixture of 3,6-di-tert-butyl-9H-carbazole (5.0 g, 17.8 mmol), tris(4-bromophenyl)amine (10.4 g, 21.4 mmol), copper(I)iodide (2.1 g, 11.3 mmol), 1,10-phenanthroline (4.2 g, 23.2 mmol) and Cs2CO3 (17.5 g, 53.7 mmol) was dissolved in 80 mL of dry DMF and refluxed under nitrogen atmosphere for 12 h. After cooling to room temperature, solvent was removed under vacuum and the residue was dissolved in chloroform. The solution was passed through celite pad to remove inorganic impurities and the filtrate was extracted with chloroform. The combined organic layer was washed with water, brine and dried over anhydrous Na2SO4. The solution was filtered, concentrated and subjected to column chromatography on silica gel with n-hexane as the eluent, to get white solid (6.9 g, 10.1 mmol, 47%). 1H NMR (500 MHz, CDCl3): δ 8.14–8.13 (d, 2H, J = 1.5 Hz), 7.49–7.35 (m, 11H), 7.33–7.23 (d, 2H, J = 10.6 Hz), 7.21–7.03 (d, 3H, J = 8.7 Hz), 1.46 (s, 18H); 13C NMR (125 MHz, CDCl3): δ 145.9, 145.2, 142.6, 138.9, 138.2, 132.9, 132.3, 132.1, 127.4, 125.6, 125.3, 124.6, 123.2, 123.0, 115.9, 115.8, 108.9, 34.5, 31.8. MS (EI): m/z 680 (M+).
4-(Benzo[c][1,2,5]thiadiazol-5-yl)-N,N-bis(4-bromophenyl)aniline (6a)
5-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[c][1,2,5]thiadiazole (2 g, 7.6 mmol) and tris(4-bromophenyl)amine (4.04 g, 8.4 mmol) were dissolved in 30 mL of dry toluene. After adding 12 mL of 2 M K2CO3 solution, the reaction mixture was bubbled with N2 gas for 15 minutes and tetrakis(triphenylphosphine)palladium (90 mg, 0.001 mmol) was added to the reaction mixture, and then heated to 80 °C for 24 h. After cooling the reaction mixture to room temperature, it was poured into water, extracted with chloroform and dried over anhydrous Na2SO4. The resulting solution was filtered and concentrated by rotary evaporation. The residue obtained was purified by column chromatography over silica gel with n-hexane as eluent to give the desired product as yellow solid (3.2 g, 5.95 mmol, 71%). 1H NMR (500 MHz, CDCl3): δ 8.12 (s, 1H), 8.04–8.01 (d, 1H, J = 9.1 Hz), 7.87–7.86 (d, 1H, J = 7.6 Hz), 7.84–7.83 (d, 2H, J = 8.3 Hz), 7.60–7.58 (d, 4H, J = 9.1 Hz), 7.40–7.37 (d, 2H, J = 8.3 Hz), 7.17–6.99 (d, 4H, J = 9.1 Hz); 13C NMR (125 MHz, CDCl3): δ 155.4, 154.0, 147.2, 145.9, 141.5, 133.9, 132.5, 129.8, 128.4, 125.9, 123.9, 121.4, 117.6, 116.2. MS (EI): m/z 537 (M+).
4-([1,2,5]Thiadiazolo[3,4-b]pyridin-6-yl)-N,N-bis(4-bromo phenyl)aniline (6b)
This compound was synthesized according to the procedure similar to that of 6a, using 6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-[1,2,5]thiadiazolo[3,4-b]pyridine (2.0 g, 7.6 mmol) to give the desired product as orange solid. The obtained product was recrystallized from CH2Cl2/hexane to get orange needles (2.7 g, 5.01 mmol, 61%). 1H NMR (500 MHz, CDCl3): δ 9.35 (s, 1H), 8.38 (s, 1H), 7.62–7.59 (d, 2H, J = 8.7 Hz), 7.43–7.40 (d, 4H, J = 8.9 Hz), 7.21–7.19 (d, 2H, J = 8.5 Hz), 7.04–7.02 (d, 4H, J = 8.7 Hz); 13C NMR (125 MHz, CDCl3): δ 160.9, 156.0, 147.9, 147.8, 145.8, 136.4, 132.6, 130.2, 128.6, 126.2, 124.8, 123.6, 116.6. MS (EI): m/z 538 (M+).
4-(Benzo[c][1,2,5]thiadiazol-5-yl)-N-(4-(benzo[c][1,2,5]thiadiazol-5-yl)phenyl)-N-(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)aniline (CTDB)
4-Bromo-N-(4-bromophenyl)-N-(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)aniline (1 g, 1.5 mmol), 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[c][1,2,5]thiadiazole (847 mg, 3.2 mmol) were dissolved in 15 mL of toluene. After adding 8 mL of 2 M K2CO3 solution, reaction mixture was bubbled with nitrogen for 15 minutes. Tetrakis(triphenylphosphine)palladium (90 mg, 0.001 mmol) was added to the reaction mixture and then heated to 80 °C for 24 h. After cooling the reaction mixture to room temperature, it was filtered through celite pad to remove the inorganic impurities and the filtrate was extracted with chloroform. The combined organic layer was washed with water, brine and dried over anhydrous Na2SO4. The resulting solution was filtered and concentrated by rotary evaporation and the residue obtained was purified by column chromatography over silica gel with n-hexane as the eluent to give the desired product as yellow solid (652 mg, 0.82 mmol, yield 55%). 1H NMR (500 MHz, CDCl3): δ 8.18–8.17 (dd, 4H, J = 9.6 Hz), 8.16–8.15 (d, 2H, J = 9.2 Hz), 8.07–8.04 (d, 4H, J = 1.7 Hz), 7.93–7.92 (d, 4H, J = 1.7 Hz), 7.91–7.69 (m, 4H), 7.53–7.37 (m, 8H), 1.48 (s, 18H); 13C NMR (125 MHz, CDCl3): δ 155.5, 154.1, 147.5, 145.5, 142.8, 141.6, 139.2, 134.0, 133.5, 129.8, 129.3, 128.5, 127.6, 125.6, 124.5, 123.5, 123.3, 121.5, 117.7, 116.3, 109.1, 34.7, 31.9. MS (MALDI-TOF): (m/z) calcd: 791.29; found: 791.51.
4-([1,2,5]Thiadiazolo[3,4-b]pyridin-6-yl)-N-(4-([1,2,5]thiadiazole[3,4-b]pyridin-6-yl)phenyl)-N-(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)aniline (CTDP)
This compound was synthesized according to the procedure similar to that of CTDB, using 6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)[1,2,5]thiadiazolo[3,4-b]pyridine (853 mg, 3.2 mmol), to give the desired product as red solid. The obtained product was recrystallized from toluene/methanol mixture to give red needles (680 mg, 0.86 mmol, 55%). 1H NMR (500 MHz, CDCl3): δ 9.42–9.41 (d, 2H, J = 2.3 Hz), 8.45 (d, 2H, J = 2.3 Hz), 8.20 (s, 2H), 7.74–7.71 (d, 4H, J = 8.5 Hz), 7.55–7.42 (m, 12H), 1.48 (s, 18H); 13C NMR (125 MHz, CDCl3): δ 160.9, 156.1, 148.1, 147.8, 145.0, 142.9, 139.1, 136.5, 134.2, 130.7, 128.8, 127.8, 126.2, 125.0, 124.5, 123.6, 123.3, 116.3, 109.1, 34.7, 31.9. MS (MALDI-TOF): (m/z) calcd: 792.28; found: 792.59.
4-(Benzo[c][1,2,5]thiadiazol-5-yl)-N,N-bis(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)aniline (DCTB)
A mixture of 3,6-di-tert-butyl-9H-carbazole (1.2 g, 4.1 mmol), 4-(benzo[c][1,2,5]thiadiazol-5-yl)-N,N-bis(4-bromophenyl)aniline (1 g, 1.9 mmol), copper(I)iodide (223 mg, 1.2 mmol), 1,10-phenanthroline (436 mg, 2.41 mmol) and Cs2CO3 (1.8 g, 5.6 mmol) was dissolved in 20 mL of dried DMF and refluxed under nitrogen atmosphere for 12 h. After cooling to room temperature, the solvent was removed under vacuum and the residue was dissolved in chloroform. The chloroform solution was filtered through celite pad to remove inorganic impurities and the filtrate was extracted with chloroform. The combined organic layers are washed with water, brine and dried over anhydrous Na2SO4. The resulting solution was filtered and concentrated by rotary evaporation. The residue was subjected to column chromatography on silica gel with n-hexane as the eluent to the desired product get yellow solid (1.2 g, 1.28 mmol, 67%). 1H NMR (500 MHz, CDCl3): δ 8.18–8.16 (m, 5H), 8.04–8.03 (d, 1H, J = 9.2 Hz), 7.91–7.89 (d, 1H, J = 9.2 Hz), 7.71–7.69 (d, 2H, J = 8.4 Hz), 7.54–7.39 (m, 20H), 1.48 (s, 36H), 13C NMR (125 MHz, CDCl3): δ 155.6, 154.2, 147.9, 145.9, 142.9, 141.8, 139.4, 133.9, 133.5, 129.9, 128.7, 127.8, 125.5, 124.3, 123.7, 123.4, 121.6, 117.8, 116.4, 109.3, 109.3, 34.8, 32.1. MS (MALDI-TOF): (m/z) calcd: 933.48; found: 933.75.
4-([1,2,5]Thiadiazolo[3,4-b]pyridin-6-yl)-N,N-bis(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)aniline (DCTP)
This compound was synthesized according to the procedure similar to that of DCTB, using 4-([1,2,5]thiadiazolo[3,4-b]pyridin-6-yl)-N,N-bis(4-bromophenyl)aniline (1 g, 1.9 mmol), to give the desired product as orange solid (0.78 g, 0.83 mmol, yield 44%). 1H NMR (500 MHz, CDCl3): δ 9.41–9.39 (d, 1H, J = 2.3 Hz), 8.42–8.14 (d, 1H, J = 2.3 Hz), 8.17 (s, 4H), 7.72–7.69 (d, 2H, J = 9.1 Hz), 7.58–7.42 (m, 16H), 7.24–7.14 (m, 2H), 1.48 (s, 36H); 13C NMR (125 MHz, CDCl3): 160.9, 156.1, 148.6, 147.8, 145.5, 142.9, 139.2, 136.6, 136.6, 133.8, 130.0, 128.9, 128.7, 128.2, 127.8, 125.8, 125.2, 124.8, 123.9, 123.6, 123.3, 116.3, 109.2, 34.7, 32.0. MS (MALDI-TOF): (m/z) calcd: 934.47; found: 934.80.
Acknowledgements
MP thanks UGC, India for the fellowship. We gratefully acknowledge CSIR for TAPSUN, NWP0055 project funding.
References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- W. Xiang, A. Gupta, M. K. Kashif, N. Duffy, A. Bilic, R. A. Evans, L. Spiccia and U. Bach, ChemSusChem, 2013, 6, 256–260 CrossRef CAS PubMed.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- Organic Photovoltaics: Materials, Device Physics, and Manufacturing Technologies, ed. U. S. Christoph Brabec and Vladimir Dyakonov, 2nd edn, 2014 Search PubMed.
- A. Gupta, A. Ali, A. Bilic, M. Gao, K. Hegedus, B. Singh, S. E. Watkins, G. J. Wilson, U. Bach and R. A. Evans, Chem. Commun., 2012, 48, 1889–1891 RSC.
- R. J. Kumar, Q. I. Churches, J. Subbiah, A. Gupta, A. Ali, R. A. Evans and A. B. Holmes, Chem. Commun., 2013, 49, 6552–6554 RSC.
- http://www.heliatek.com/en/press/press-releases/details/heliatek-consolidates-its-technology-leadership-by-establishing-a-new-world-record-for-organic-solar-technology-with-a-cell-effi.
- J. E. Coughlin, Z. B. Henson, G. C. Welch and G. C. Bazan, Acc. Chem. Res., 2013, 47, 257–270 CrossRef PubMed.
- Z. B. Henson, K. Mullen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS PubMed.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2007, 130, 732–742 CrossRef PubMed.
- Y. Chen, X. Wan and G. Long, Acc. Chem. Res., 2013, 46, 2645–2655 CrossRef CAS PubMed.
- Q. Shi, P. Cheng, Y. Li and X. Zhan, Adv. Energy Mater., 2012, 2, 63–67 CrossRef CAS.
- L. Xue, J. He, X. Gu, Z. Yang, B. Xu and W. Tian, J. Phys. Chem. C, 2009, 113, 12911–12917 CAS.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. J. Fréchet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS PubMed.
- J. Zhou, Y. Zuo, X. Wan, G. Long, Q. Zhang, W. Ni, Y. Liu, Z. Li, G. He, C. Li, B. Kan, M. Li and Y. Chen, J. Am. Chem. Soc., 2013, 135, 8484–8487 CrossRef CAS PubMed.
- J. Zhou, X. Wan, Y. Liu, Y. Zuo, Z. Li, G. He, G. Long, W. Ni, C. Li, X. Su and Y. Chen, J. Am. Chem. Soc., 2012, 134, 16345–16351 CrossRef CAS PubMed.
- L. Zhang, S. Zeng, L. Yin, C. Ji, K. Li, Y. Li and Y. Wang, New J. Chem., 2013, 37, 632–639 RSC.
- S. Steinberger, A. Mishra, E. Reinold, J. Levichkov, C. Uhrich, M. Pfeiffer and P. Bauerle, Chem. Commun., 2011, 47, 1982–1984 RSC.
- S. Steinberger, A. Mishra, E. Reinold, C. M. Müller, C. Uhrich, M. Pfeiffer and P. Bäuerle, Org. Lett., 2010, 13, 90–93 CrossRef PubMed.
- J. Zhou, X. Wan, Y. Liu, G. Long, F. Wang, Z. Li, Y. Zuo, C. Li and Y. Chen, Chem. Mater., 2011, 23, 4666–4668 CrossRef CAS.
- Y.-H. Chen, L.-Y. Lin, C.-W. Lu, F. Lin, Z.-Y. Huang, H.-W. Lin, P.-H. Wang, Y.-H. Liu, K.-T. Wong, J. Wen, D. J. Miller and S. B. Darling, J. Am. Chem. Soc., 2012, 134, 13616–13623 CrossRef CAS PubMed.
- S.-W. Chiu, L.-Y. Lin, H.-W. Lin, Y.-H. Chen, Z.-Y. Huang, Y.-T. Lin, F. Lin, Y.-H. Liu and K.-T. Wong, Chem. Commun., 2012, 48, 1857–1859 RSC.
- L.-Y. Lin, Y.-H. Chen, Z.-Y. Huang, H.-W. Lin, S.-H. Chou, F. Lin, C.-W. Chen, Y.-H. Liu and K.-T. Wong, J. Am. Chem. Soc., 2011, 133, 15822–15825 CrossRef CAS PubMed.
- D. Demeter, V. Jeux, P. Leriche, P. Blanchard, Y. Olivier, J. Cornil, R. Po and J. Roncali, Adv. Funct. Mater., 2013, 23, 4854–4861 CAS.
- D. Sahu, C.-H. Tsai, H.-Y. Wei, K.-C. Ho, F.-C. Chang and C.-W. Chu, J. Mater. Chem., 2012, 22, 7945–7953 RSC.
- A. Cravino, S. Roquet, P. Leriche, O. Aleveque, P. Frere and J. Roncali, Chem. Commun., 2006, 13, 1416–1418 RSC.
- H. Shang, H. Fan, Y. Liu, W. Hu, Y. Li and X. Zhan, Adv. Mater., 2011, 23, 1554–1557 CrossRef CAS PubMed.
- Z. Lu, C. Li, T. Fang, G. Li and Z. Bo, J. Mater. Chem. A, 2013, 1, 7657–7665 CAS.
- D. Patra, C.-C. Chiang, W.-A. Chen, K.-H. Wei, M.-C. Wu and C.-W. Chu, J. Mater. Chem. A, 2013, 1, 7767–7774 CAS.
- X. Sun, Y. Zhou, W. Wu, Y. Liu, W. Tian, G. Yu, W. Qiu, S. Chen and D. Zhu, J. Phys. Chem. B, 2006, 110, 7702–7707 CrossRef CAS PubMed.
- Y. Sun, S.-C. Chien, H.-L. Yip, Y. Zhang, K.-S. Chen, D. F. Zeigler, F.-C. Chen, B. Lin and A. K. Y. Jen, J. Mater. Chem., 2011, 21, 13247–13255 RSC.
- L. Ying, B. B. Y. Hsu, H. Zhan, G. C. Welch, P. Zalar, L. A. Perez, E. J. Kramer, T.-Q. Nguyen, A. J. Heeger, W.-Y. Wong and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 18538–18541 CrossRef CAS PubMed.
- S. Chaurasia, C.-Y. Hsu, H.-H. Chou and J. T. Lin, Org. Electron., 2014, 15, 378–390 CrossRef CAS.
- G. C. Welch and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 4632–4644 CrossRef CAS PubMed.
- X. Liu, Y. Sun, L. A. Perez, W. Wen, M. F. Toney, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2012, 134, 20609–20612 CrossRef CAS PubMed.
- K. Takimiya, I. Osaka and M. Nakano, Chem. Mater., 2013, 26, 587–593 CrossRef.
- J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martnez-Ferrero, E. Palomares, J. Ko, M. Gratzel and T. Torres, Angew. Chem., 2007, 119, 8510–8514 CrossRef.
- P. Moonsin, N. Prachumrak, R. Rattanawan, T. Keawin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2012, 48, 3382–3384 RSC.
- G.-G. Shan, H.-B. Li, J.-S. Qin, D.-X. Zhu, Y. Liao and Z.-M. Su, Dalton Trans., 2012, 41, 9590–9593 RSC.
- M. Paramasivam, R. K. Chitumalla, S. P. Singh, A. Islam, L. Han, V. Jayathirtha Rao and K. Bhanuprakash, J. Phys. Chem. C, 2015, 119, 17053–17064 CAS.
- A. Baheti, P. Tyagi, K. R. J. Thomas, Y.-C. Hsu and J. T. Lin, J. Phys. Chem. C, 2009, 113, 8541–8547 CAS.
- V. R. Gopal, A. M. Reddy and V. J. Rao, J. Org. Chem., 1995, 60, 7966–7973 CrossRef CAS.
- V. Raj Gopal, V. Jayathirtha Rao, G. Saroja and A. Samanta, Chem. Phys. Lett., 1997, 270, 593–598 CrossRef CAS.
- U. Srinivas, P. Arun Kumar, K. Srinivas, K. Bhanuprakash and V. Jayathirtha Rao, J. Struct. Chem., 2012, 53, 851–865 CrossRef CAS.
- S. Fantacci, F. De Angelis, A. Sgamellotti and N. Re, Chem. Phys. Lett., 2004, 396, 43–48 CrossRef CAS.
- M. Paramasivam, A. Gupta, A. M. Raynor, S. V. Bhosale, K. Bhanuprakash and V. Jayathirtha Rao, RSC Adv., 2014, 4, 35318–35331 RSC.
- K. R. J. Thomas, N. Kapoor, M. N. K. P. Bolisetty, J.-H. Jou, Y.-L. Chen and Y.-C. Jou, J. Org. Chem., 2012, 77, 3921–3932 CrossRef CAS PubMed.
- S. Roquet, A. Cravino, P. Leriche, O. Alévêque, P. Frère and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459–3466 CrossRef CAS PubMed.
- P. Leriche, P. Frère, A. Cravino, O. Alévêque and J. Roncali, J. Org. Chem., 2007, 72, 8332–8336 CrossRef CAS PubMed.
- D. Demeter, T. Rousseau, P. Leriche, T. Cauchy, R. Po and J. Roncali, Adv. Funct. Mater., 2011, 21, 4379–4387 CrossRef CAS.
- A. Cravino, P. Leriche, O. Alévêque, S. Roquet and J. Roncali, Adv. Mater., 2006, 18, 3033–3037 CrossRef CAS.
- J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS PubMed.
- T.-T. Bui, L. Beouch, X. Sallenave and F. Goubard, Tetrahedron Lett., 2013, 54, 4277–4280 CrossRef CAS.
- N. Prachumrak, S. Pansay, S. Namuangruk, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Eur. J. Org. Chem., 2013, 2013, 6619–6628 CrossRef CAS.
- M. Sonntag, K. Kreger, D. Hanft, P. Strohriegl, S. Setayesh and D. de Leeuw, Chem. Mater., 2005, 17, 3031–3039 CrossRef CAS.
- M. A. Reddy, A. Thomas, K. Srinivas, V. J. Rao, K. Bhanuprakash, B. Sridhar, A. Kumar, M. N. Kamalasanan and R. Srivastava, J. Mater. Chem., 2009, 19, 6172–6184 RSC.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 1996–2000 CrossRef CAS PubMed.
- V. S. Bryantsev, M. S. Diallo, A. C. T. van Duin and W. A. Goddard, J. Chem. Theory Comput., 2009, 5, 1016–1026 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of NMR and mass spectral data, absorption and emission profiles of the dyes, X-ray crystallographic data of intermediate 6b. CCDC 1038010. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra06616j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 








