
Influence of support on the performance of copper catalysts for the effective hydrogenation of ethylene carbonate to synthesize ethylene glycol and methanol
Abstract
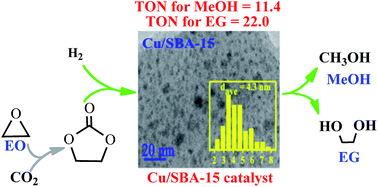
Hydrogenation of ethylene carbonate (EC) is an attractive and promising approach for indirect hydrogenation of CO2 to co-produce ethylene glycol (EG) and methanol (MeOH). In this work, thermodynamics of EC hydrogenation was firstly calculated, and the results disclosed that EC hydrogenation is exothermic (ΔrHθm = −71.59 kJ mol−1) and thermodynamically favorable (ΔrGθm = −25.62 kJ mol−1). The mesoporous silica supported copper catalysts were successfully prepared by facile ammonia evaporation method using KIT-6, MCM-41 and SBA-15 as support materials. The as-prepared Cu/KIT-6, Cu/MCM-41 and Cu/SBA-15 were thoroughly characterized by N2 physisorption, ICP-AES, N2O titration, FT-IR, XRD, H2-TPR, TEM, XPS and XAES. It was found that copper nanoparticles were well dispersed on these supports. Interestingly, a larger metallic Cu surface area with higher dispersion was obtained on SBA-15 when compared with KIT-6 and MCM-41. Furthermore, Cu0 and Cu+ species in various ratios were verified to co-exist on the surfaces of the three catalysts, which originated from CuO and copper phyllosilicate, respectively. The catalytic performances showed that Cu/SBA-15 exhibited a superior catalytic activity with a TON of 22.0 and 11.4 (mol mol−1) towards EG and MeOH, respectively. Importantly, 100% of EC conversion, 94.7% of EG yield and 62.3% of MeOH yield were achieved within a short reaction time of 4 h over Cu/SBA-15 under optimized conditions. The synergetic effect between Cu0 and Cu+ species, in which it was proposed that Cu0 species dissociated H2, while Cu+ species absorbed the carbonyl group of EC, was responsible for the higher catalytic activity of Cu/SBA-15.
 Please wait while we load your content...
Please wait while we load your content...

