
Synthesis of photo-responsive azobenzene molecules with different hydrophobic chain length for controlling foam stability†
Abstract
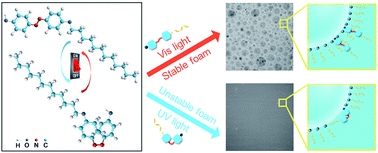
To obtain an aqueous foam photo-switch, azobenzene molecules [4-hydroxy-4′-oxoalkyl azobenzene (HCnAzo) n = 4, 8, and 12] were synthesized. The hydrophobic chain length affected both photo-responsive properties and foam stability controllability. trans → cis isomerization of HCnAzo occurred by exposing to UV light for 1 s and the cis → trans process for HC4Azo, HC8Azo and HC12Azo was carried out by visible light irradiation for 12 min, 13 min and 14 min, respectively. The reversible isomerization was repeatable, maintaining high sensitivity. Due to the small steric effect and isomerization barrier, photo-isomerization of HC4Azo was more rapid than HC8Azo and HC12Azo. With the increase of the hydrophobic chain length, the decrease of thermal isomerization barrier resulted in less cis isomer and more trans isomer in photo-stationary state via UV and visible light irradiation, respectively. The combination of visible light and heat can accelerate the cis → trans isomerization speed. trans HC4Azo, HC8Azo and HC12Azo increased foam life from 6.67 min to 10.38, 9.91 and 7.74 min, respectively, resulting from the absorption on the air–water interface and a high affinity. cis HC4Azo, HC8Azo and HC12Azo decreased foam life from 6.67 min to 5.12, 5.49 and 6.02 min, respectively, attributed to the interfacial desorption and increase of surface tension. HC4Azo with sensitive photo-isomerization and effective foam stability controllability was the best choice for an aqueous foam photo-switch.
 Please wait while we load your content...
Please wait while we load your content...

