DOI:
10.1039/C6RA06412D
(Paper)
RSC Adv., 2016,
6, 52310-52317
Novel Zn(II)-thiazolone-based solid fluorescent chemosensors: naked-eye detection for acid/base and toluene†
Received
10th March 2016
, Accepted 13th May 2016
First published on 19th May 2016
Abstract
A series of novel thiazolone-based zinc complexes, [Zn(Am4DHotaz)Cl2]·H2O (1), [Zn(Am4Motaz)Cl2] (2), and [Zn(Am4Eotaz)Cl2] (3) (Am4DHotaz = N-(4-oxothiazolidin-2-ylidene)picolinohydrazonamide, Am4Motaz = N-(3-methyl-4-oxothiazolidin-2-ylidene)picolinohydrazonamide, Am4Eotaz = N-(3-ethyl-4-oxothiazolidin-2-ylidene)picolinohydrazonamide), with intense fluorescence emission and large Stokes shifts in the solid state, have been synthesized and characterized by X-ray crystallography and various spectroscopic methods. Complexes 1–3 exhibit a similar Zn2+-coordinated pattern, but different molecular packings arising from substituent variation in the organic ligands, which finally result in individual fluorescent behaviors. All the complexes showed interesting ON/OFF/ON fluorescence switching properties induced by acid/base vapor. More interestingly, for 1, the introduction of toluene brought about a large blue shift, an obvious color change suitable for naked-eye detection, and a distinct increase of fluorescent quantum yield, which could contribute to environmental monitoring and other applications in the field of luminescent materials.
Introduction
The current enormous interest in exploiting metal complexes as fluorescent sensors stems not only from their potential promising optical spectrum properties but also from their various application prospects in life science and environmental fields.1,2 And it has become one of the foremost topics in modern coordination chemistry.3,4 So far, many fluorescent metal complexes based on coumarin, rhodamine, BODIPY, fluorescein, quinoline, naphthalimide, and others have been reported.5–11 However, most of these probes have some drawbacks, such as complicated synthesis, small Stokes shifts, or a self-quenching tendency in solid state, some of which could limit their practical applications in photoelectric conversion and electroluminescence materials.12–14 Thus the improved generation presents a significant challenge.
Zinc complex with highly efficient fluorophore is one of the most important fluorescent sensors.15,16 It is worth noting that many reported fluorescent “turn-on” chemosensors employ zinc(II) as a central ion due to its advantageous photophysical properties.17,18 For small Zn(II)-fluorophore molecules, packing manners and donor–acceptor (D–A) architectures are critical for the determination of electron-transporting properties.19,20 Molecular packing can be greatly affected by many intermolecular factors, such as π–π stacking and hydrogen bonds.21,22 Therefore, optimization of ligand structures is required in designing such fluorescent probes, in order to alleviate fluorescence quenching caused by π–π packing and promote an efficient intramolecular charge transfer (ICT) process that enhances the Stokes shift.
Toluene is an aromatic hydrocarbon used in numerous products, including paints, adhesives, inks etc., which are largely applied in the automobile and shoemaking industries.23,24 The highest levels of toluene discharging and exposure arise in the industries listed above and may severely impact the life of people around those areas.25 Acute and chronic intake is confirmed to cause multiple effects in the body, such as hurt to the central nervous, hematopoietic, cardiovascular, renal, immune and gastrointestinal systems.26–31 It also produces severe impairments of electrolyte and pH balance, even generates carcinogens.32,33 Several methods, for example, gas chromatography and spectrophotometry34,35 have been reported for the detection of toluene. Although these methods are capable of detecting toluene, they are too complicated, uneconomic and time-consuming. As far as we know, the existing reports on toluene detection are relatively rare, especially through chemical sensor analysis. More importantly, among various sensing systems, naked-eye-response probes, which could provide a simple and economical means to detect the targeted analyte without sophisticated instrumentation, have been of considerable interest.36 Therefore, it is everlastingly desirable to explore facile and inexpensive fluorescence sensors for toluene detection.
Here we present three novel convenient and effective Zn-type sensors, in which the chemical groups with low electron density, such as pyridine, were employed to coordinate with Zn2+ to form strong D–A moieties with deep LUMO (Lowest Unoccupied Molecular Orbital) levels. Following Am4DHotaz and Am4Motaz, which were first described by Alfonso Castiñeiras group in 2008,37 a new ligand, Am4Eotaz, with ethyl instead of hydrogen or methyl, was introduced in this work. Three ligands were obtained by a two-step synthesis. Consequently, three novel thiazolone-based zinc fluorescent molecules, [Zn(Am4DHotaz)Cl2]·H2O (1), [Zn(Am4Motaz)Cl2] (2), and [Zn(Am4Eotaz)Cl2] (3), were synthesized and characterized by X-ray crystallography and various spectroscopic methods. All synthetic routes of ligands and complexes 1–3 are illustrated in Scheme S1.† These molecules with different substituents of ligands, packing patterns, and donor–acceptor architectures, displayed diverse colour with high fluorescence quantum yields and large Stokes shifts. In particular, complex 1 could selectively detect toluene with naked eye.
Experimental
Materials and physical measurements
All reagents were obtained from commercial sources. All chemical solvents were dried and distilled by standard methods before use (cautious: MeOH must dry over Mg and I2). IR spectra were recorded on a Nicolet 380 spectrometer as KBr pellets in the range 4000–375 cm−1 with the OMNIC software. NMR measurements were recorded on a Bruker AVANCE III 400 spectrometer with tetramethylsilane as internal standard. Single crystals data were collected on a Rigaku Saturn X-ray CCD diffractometer using MoKα radiation. Elemental analysis for C, H and N was performed on a Perkin-Elmer analyzer model 240. UV spectra were measured using a JASCO V-770 spectrophotometer. Fluorescence spectral data were obtained using a Shimadzu RF-5301 fluorescence spectrophotometer at the approved temperature. The absolute fluorescence quantum yields were determined on an Edinburgh FLS920 fluorescence spectrometer in an integrating sphere.
Preparation of 1,3-thiazolidin-4-ones (ligands)
The ligands were synthesized according to a procedure reported previously.37,38 2-Cyanopyridine was dissolved in sodium methoxide solution under stirring for 0.5 h. The thiosemicarbazide was added in small portions to the above solution, the mixture was then refluxed for another 4 h. The pale yellow solution was cooled to room temperature and filtered, after that, the yellowish precipitation was dissolved and recrystallized from methanol. The product and several drops of triethylamine were mixed in toluene solvent, after stirring 10 min, 1.2 equivalent of chloroacetic acid was added. The suspension was refluxed for 2 h to give the precipitate which was filtered off and washed with toluene for several times. The precipitate was recrystallized from ethanol to give pure white Am4DHotaz (yield: ca. 69%), yellowish Am4Motaz (yield: ca. 60.3%) and yellowish Am4Eotaz (yield: ca. 69%) respectively. IR, 1H and 13C NMR analysis data of these ligands were listed in ESI.†
Preparation of complexes
Preparation of [Zn(Am4DHotaz)Cl2]·H2O (1). A 5 mL MeOH solution of ZnCl2·6H2O (0.049 g, 0.2 mmol) was added into a 10 mL yellowish solution of Am4DHotaz (0.047 g, 0.2 mmol) under stirring for 3 h. The resulting yellow suspension was filtered, and the filtrate was left to stand at room temperature for slow evaporation. The light yellow block crystals suitable for X-ray analysis were collected after several days. Yield: 56 mg (ca. 59% based on Zn). Elemental analysis (%): calc. for C9H9Cl2ZnN5OS: C, 29.09; H, 2.44; N, 18.85. Found: C, 28.95; H, 2.58; N, 18.69. Selected IR data (KBr, cm−1): 3383, 3296.4, 3052.5, 2968.1, 2925.5, 1654.1, 1589.8, 1571.9, 1514.1, 1472.2, 1398.1, 1343.6, 1246.2, 1198, 795.1, 655.1, 634.1.
Preparation of [Zn(Am4Motaz)Cl2] (2). Complex 2 was prepared according to the similar procedure of 1, using Am4Motaz (0.050 g, 0.2 mmol) instead of Am4DHotaz (0.047 g, 0.2 mmol). Yellow block crystals of 2 suitable for X-ray analysis were collected from the final filtrate. Yield: 45 mg (ca. 46% based on Zn). Elemental analysis (%): calc. for C10H11Cl2ZnN5OS: C, 31.15; H, 2.88; N, 18.16. Found: C, 30.98; H, 3.05; N, 18.06. Selected IR data (KBr, cm−1): 3414.6, 3309.2, 3264.2, 1717.4, 1639.6, 1614.6, 1589.1, 1486.1, 1423.8, 1392.5, 1365.1, 1315.8, 1230, 1120, 1026.8, 889.8, 535.2, 490.2.
Preparation of [Zn(Am4Eotaz)Cl2] (3). Complex 3 was prepared according to the similar procedure of 1, using Am4Eotaz (0.053 g, 0.2 mmol) instead of Am4DHotaz (0.047 g, 0.2 mmol). Yellow block crystals of 3 suitable for X-ray analysis were collected from the final filtrate. Yield: 53 mg (ca. 52% based on Zn). Elemental analysis (%): calc. for C11H13Cl2ZnN5OS: C, 33.06; H, 3.28; N, 17.53. Found: C, 32.92; H, 3.35; N, 17.36. Selected IR data (KBr, cm−1): 3396.1, 3263.5, 3176.9, 3080.5, 2978, 1706.1, 1644.6, 1613.3, 1590.8, 1487.8, 1422.1, 1390.1, 1341, 1301.8, 1248, 1132, 1100.4, 1054.4, 1024.7, 889.6, 801.6, 775.3, 724.4, 632.1, 498.
X-ray crystallography
Diffraction data for 1–3 were collected at 113(2) K with a Rigaku Saturn CCD diffractomer equipped with graphite monochromated MoKα radiation by using the ω-scan technique. The data were processed using CrystalClear software39 and corrected for Lorentz and polarization effects. Absorption corrections were applied by using a multiscan program. The structures were solved by direct methods and refined by full-matrix least squares based on F2 using the SHELXTL program package.40 Non-hydrogen atoms were subjected to anisotropic refinement. Hydrogen atoms were assigned with common isotropic displacement factors. Hydrogen atoms were included at geometrically calculated positions and refined using a riding model except those bonded to the oxygen atoms in water molecules, which were located on a different Fourier map. Crystallographic data and experimental details for structural analyses are summarized in Table S1,† and selected bond lengths (Å) and bond angles (°) of 1–3 in Table S2.† Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, CCDC no. 1433220, 1433094, and 1433095 for 1–3.
Photoluminescence experiments
Crystals of 1–3 were grinded into powder in order to perform UV, fluorescence, and quantum yield measurements. Each of the powdered samples was exposed to HCl and TEA (triethylamine) vapor alternately for each 15 min to achieve the “ON/OFF/ON” fluorescence switching experiment. In the toluene-induced experiment, powder 1 was added into toluene solution (methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene = 9:1) and sonicated, then the obtained powder was used to perform further analysis.
:toluene = 9:1) and sonicated, then the obtained powder was used to perform further analysis.
Theoretical calculations
Theoretical calculations for 1–3 were performed on density-functional theory (DFT) level as implemented in the Gaussian 09 suit of programs.41 Becke's three-parameter nonlocal exchange functional along with the Lee–Yang–Parr nonlocal correlation functional (B3LYP) was employed. The LANL2DZ basis set was used for Zn atom, and the 6-31+G(d) was used for other atoms.
Results and discussion
Description of syntheses and crystal structures of 1–3
The three complexes were obtained by an analogous route in which Am4Dhotaz, Am4Motaz and Am4Eotaz were employed as chelating ligands to react with ZnCl2·6H2O in MeOH, respectively. Block-shaped crystals suitable for X-ray analysis were obtained by slow solvent evaporation after several days. Single-crystal X-ray diffraction analyses revealed that complexes 1–3 crystallized in the monoclinic system with space group P21/c, P21/c and P21/n, respectively, showing extremely similar neutral mononuclear coordination structures as shown in Fig. 1. The Zn(II) center in each of the three complexes lies in a distorted tetrahedron coordination environment, defined by two nitrogen atoms from ligands and two Cl− anions. The Zn–N bond distances lie in the range of 2.0606(15)–2.074(3) Å, while the Zn–Cl bond distances vary in that of 2.2095(6)–2.2323(10) Å. There is one lattice water molecule in 1, and nothing in 2 and 3. However, the packing patterns of mononuclear units in 1–3 are significantly different, arising from the introduction of methyl/ethyl substituents on the thiazole ring for 2 and 3, respectively. For 1, adjacent mononuclear units are linked by N–H⋯O hydrogen bonds between thiazole rings and carbonyl oxygen atoms, while N–H⋯Cl hydrogen bonds between amine groups and chlorine atoms generate infinite 2D planes (Fig. 2 and Table 1). Uncoordinated water molecules act as space filling particles here and form intricate interplanar hydrogen bonds with carbonyl oxygen atoms or amine groups from ligands, and coordinated chlorine atoms, thus construct a 3D supramolecular structure. In addition, the opposite pyridyl rings of Am4Dhotaz ligand from neighboring planes are parallel to each other with an interplanar distance of 3.375 Å, showing the offset face-to-face π–π stacking, which contributes to the forming of 3D network together with extensive hydrogen bonds (Fig. S1† and Table 1). For 2, two coordinated Cl atoms both construct hydrogen bonds with amine groups, which forms 1D chain (Fig. 3 and Table 1). For 3, the amino group forms N–H⋯O and N–H⋯Cl hydrogen bonds with the carbonyl oxygen and chlorine atoms from adjacent mononuclear unit, forming a 2D plane (Fig. 4 and Table 1). Moreover, the π–π stacking between neighboring pyridyl rings from Am4Eotaz ligands display the interplanar distance of 3.751 Å in 3 (Fig. S2†), which is slightly longer than that of 1, and there is almost no overlap between intermolecular chromophore cores in 2.
 |
| | Fig. 1 Perspective views of molecular structures of complexes 1–3 (hydrogen atoms omitted for clarity). | |
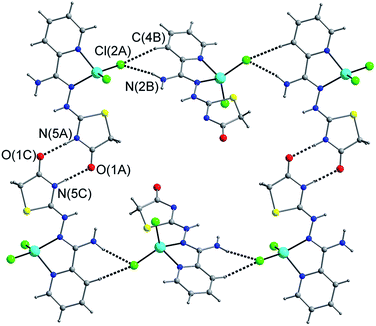 |
| | Fig. 2 The packing diagram of 1 showing 2D supramolecular network constructed by hydrogen bonds. | |
Table 1 Bond lengths [Å] and bond angles [°] of hydrogen bonds for complexes 1–3a
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
∠(DHA) |
| Symmetry transformations used to generate equivalent atoms for 1: #1 −x, −y + 1, −z; #2 x, −y + 3/2, z − 1/2; #3 −x − 1, −y + 1, −z; #4 −x, y − 1/2, −z + 1/2; #5 x + 1, y, z. For 2: #1 x, −y + 1/2, z − 1/2. For 3: #1 −x + 3/2, y + 1/2, −z + 1/2; #2 x + 1/2, −y + 1/2, z − 1/2. |
| Complex 1 |
| N2–H2B⋯O2#1 |
0.86 |
2.34 |
3.113(5) |
149.9 |
| N2–H2C⋯Cl2#2 |
0.86 |
2.44 |
3.289(3) |
167.6 |
| N5–H5A⋯O1#3 |
0.86 |
1.98 |
2.833(4) |
171.2 |
| O2–H1W⋯Cl1#4 |
0.862(10) |
2.67(3) |
3.490(4) |
158(6) |
| O2–H2W⋯O1#5 |
0.863(10) |
2.15(2) |
2.991(4) |
165(6) |
|
| Complex 2 |
| N2–H2A⋯Cl2#1 |
0.86 |
2.68 |
3.335(3) |
133.6 |
| N2–H2B⋯Cl1#1 |
0.86 |
2.52 |
3.272(3) |
146.9 |
|
| Complex 3 |
| N2–H2B⋯Cl1#1 |
0.86 |
2.89 |
3.4053(17) |
119.9 |
| N2–H2C⋯O1#2 |
0.86 |
2.04 |
2.885(2) |
66.0 |
 |
| | Fig. 3 The zigzag chain in 2 formed through hydrogen bonds. | |
 |
| | Fig. 4 Views of edge-to-face motif linked by hydrogen bonds in 3. | |
The different packing patterns of 1–3 are due to various substituent group on thiazole ring. The participations of methyl and ethyl group in 2 and 3 enhance the steric hindrance around the thiazole ring, meanwhile, they lead to different π–π stacking and hydrogen bond modes from those in 1.
Photoluminescence properties
Generally, compounds containing acyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds are usually non-fluorescent, because the CN isomerization is apt to cause free rotation which is the predominant decay process of the excited state.42–46 Inevitably, the complexes we synthesized do not show any noticeable emission in solution when excited at 300 nm, which results from the CN isomerization in molecules. None of the free ligands described above in solid states shows visible fluorescence upon irradiation with a UV lamp, however, the solid states of 1, 2 and 3 exhibit bright yellow, cyan, and spring green fluorescence, respectively, as displayed in Fig. 5.
N bonds are usually non-fluorescent, because the CN isomerization is apt to cause free rotation which is the predominant decay process of the excited state.42–46 Inevitably, the complexes we synthesized do not show any noticeable emission in solution when excited at 300 nm, which results from the CN isomerization in molecules. None of the free ligands described above in solid states shows visible fluorescence upon irradiation with a UV lamp, however, the solid states of 1, 2 and 3 exhibit bright yellow, cyan, and spring green fluorescence, respectively, as displayed in Fig. 5.
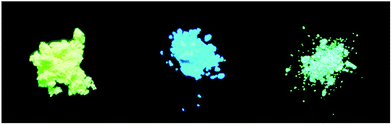 |
| | Fig. 5 Photo of solid fluorescence of 1 (left), 2 (middle) and 3 (right), taken under UV (365 nm) lamp. | |
In solid state, the major absorption peaks of 1, 2 and 3 are situated at 340, 314 and 322 nm (Table 2 and Fig. S3†), respectively, and the emissions could be observed around 500 nm upon appropriate excitation (Fig. 6). Apparently, the Stokes shifts are approximately 150 nm which are larger than related probes reported in literature, such as 54 nm for rhodamine-based chemosensor.47 The fluorescence quantum yield (Φ) increases in the order 1 (0.12) < 3 (0.15) < 2 (0.45), which is ascribable to different molecular packing modes in space.48 Thus, to explain such phenomenon, the X-ray crystal diffraction data should be taken into account. In complexes 1–3, high similarity of mononuclear units could be observed while subtle differences exist in their molecular planarity. The dihedral angles between the pyridine plane and thiazole plane are 28.18° (2) > 20.75° (3) > 16.48° (1) (Fig. S4†). Among of them, complex 2 displays the largest dihedral angle (the worst planarity), which could decrease its π–π stacking and intermolecular action in 3D space. In complex 1, the π–π overlapping was formed between the adjacent pyridine rings, and the vertical distance between the two ring centroids is 3.375 Å, while the distance between the opposite rings in 3 is 3.751 Å, slightly longer than that of 1. There is almost no overlap between intermolecular chromophore cores in 2. Thus, the different fluorescence feature is tentatively attributed to an alteration of planarity of molecules caused by introducing new substituents. The similar conclusion was reported previously in literatures.49 The π–π stacking is known to be able to cause fluorescence quenching in solid state.50 Hence, it can be supposed that 1 and 3 show a lower quantum yield. Obviously, the data of fluorescence quantum yield are in accordance with the π–π distance sequence. Furthermore, it has been known that strong and continuous intermolecular interactions between neighbouring molecules cause fluorescence quenching in the solid states. For example, Cl(2) atom forms hydrogen bond with the donor nitrogen from adjacent molecule (N–H⋯Cl, distance: 3.289(3) Å, angle: 167.6°) in 1 (Table 1). Simultaneously, N(5) and O(1) interact with the O and N atoms from neighbouring molecule to form hydrogen bonds (N5–H5A⋯O1 2.833(4) Å), respectively, and Cl(1) forms O2–H1W⋯Cl1 with O(2) of water molecule (distance: 3.490(4) Å, angle: 158°) in 1. Meanwhile, less intermolecular interactions such as hydrogen bonds can be found in 2 and 3.
Table 2 Electronic absorption, fluorescence, and photophysical data of solid samples for 1–3
| |
λabsa (nm) |
λemb (nm) |
Φfc |
SS (nm) |
| λabs: longest absorption maximum. λem: emission maximum upon excitation at the longest absorption maximum. Φf: absolute quantum yield. SS: Stokes shift. |
| 1 |
340 |
503 |
0.12 |
163 |
| 2 |
314 |
464 |
0.45 |
150 |
| 3 |
322 |
474 |
0.15 |
152 |
 |
| | Fig. 6 Fluorescence emission spectra of 1–3 in solid state. | |
As discussed above, π–π stacking at low level and few intermolecular interactions provide favorable factors that would eliminate self-quenching and enhance solid fluorescence.51,52
Theoretical calculations
For a better understanding of the optical properties displayed by 1–3, calculations were carried out with the Gaussian 09 suit of programs, using the B3LYP level. The LANL2DZ basis set was used for Zn atom and the 6-31+G(d) was used for the remaining atoms. Geometric parameters from X-ray diffraction analysis were used for the calculation. Fig. 7 shows HOMO and LUMO levels of 1–3. It illustrates that the LUMO distribution is donated by all atomic orbitals in the aromatic rings, while contributions to the HOMO distribution are mainly from part A. Therefore, efficient intramolecular charge transfer (ICT) process could be a key factor for their broad bands in absorption and large Stokes shifts.53
 |
| | Fig. 7 Diagrams of HOMO and LUMO levels of 1–3. | |
The calculated HOMO–LUMO band gaps of 1–3 are 3.12, 3.381 and 3.405 eV respectively. The added electron-donating groups in conjugation system of 2 and 3 increase the LUMO energy relatively, which in turn cause blue-shifting both in absorption and emission spectra. However, the better optical feature of 2 makes it more convinced that π–π stacking and intermolecular interactions provide crucial factors for the fluorescence intensity.
Solid-state “ON/OFF/ON” fluorescence switching
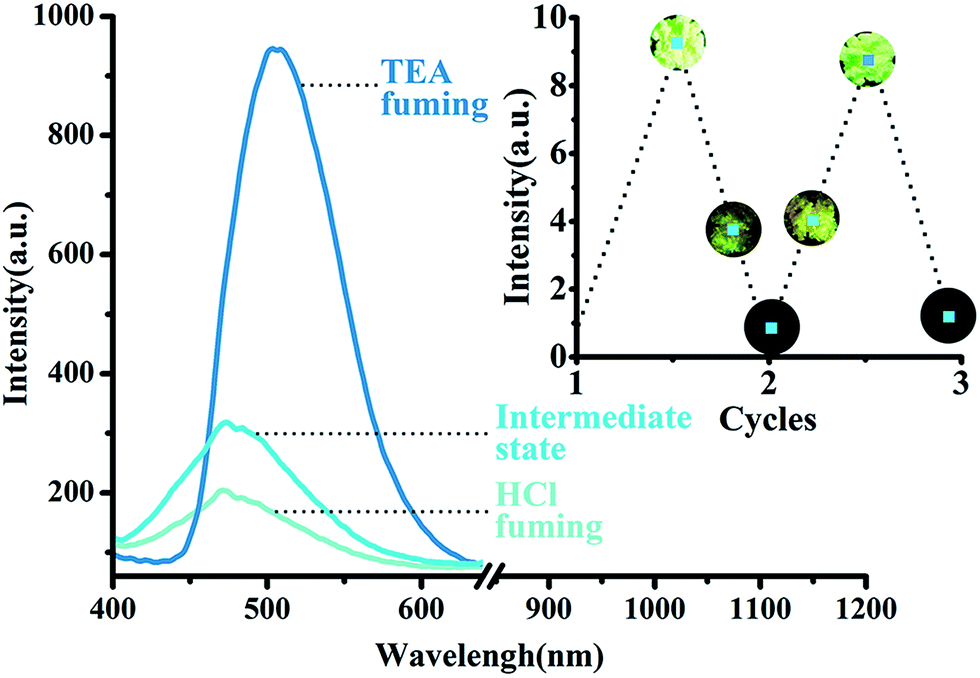
The fluorescence of solids 1–3 are sensitive to hydrochloride acid (HCl) vapor due to the Lewis-base nature of nitrogen atoms in amino groups (Fig. 8). For example, after being evaporated by HCl vapor for fifteen minutes, the yellow sample of 1 was converted into a white solid followed by fluorescence changing from bright yellow into dark as well as emission intensity quenching (indicating that the fluorescence is turned “OFF”), likely owing to the protonation of the amino group. And the quenched emission is probably due to the changed molecular conformation and packing structure.54 The white sample gradually recovered its original bright yellow fluorescence (turn “ON”) through a dark yellow intermediate state when it was treated with triethylamine vapor for another fifteen minutes. The switching between fluorescence “OFF” and “ON” states by acid/base vapor fuming can be carried out repeatedly without obvious intensity decaying (Fig. 9). Complexes 2 and 3 displayed similar switching pattern to 1 (see Fig. S8 and S9†).
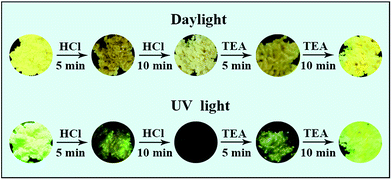 |
| | Fig. 8 Fluorescent dark and bright state of 1 switched by protonation/deprotonation process. | |
 |
| | Fig. 9 Fluorescence “ON/OFF/ON” switching of 1 performed by acid/base vapor fuming process. | |
Solid-state for selective toluene detection
The powder of 1 shows strong bright yellow luminescence on irradiation with UV light (λ = 366 nm). More interestingly, after a small amount of toluene being added, the luminescence of the powder changes into cyan with fluorescence quantum yield increasing from 0.12 to 0.56 (approximately 5 times as high as 1). For comparison, other common organic solvents, such as benzene, xylenes, naphthalene, chloroform, 1,4-dioxane, do not have this effect (Fig. S10†). The resulting fluorescence band is more blue-shifted about 40 nm than that of 1 alone (Fig. 10). This behaviour might be explained by re-establishing the rather larger separation between the molecular planes and the resulting weak overlap of the two π systems when exposed to toluene.55 Complex 1 is a striking example of a molecule whose fluorescence emission spectrum was drastically influenced by the surrounding medium, whereas 2 and 3 do not have this feature. It might be explained by the presence of excimer fluorescence in 1, and the formation of excimer could be prevented either by substituents shielding the chromophore (as in 2) or by crystallization in an unsuitable space group (as in 3).56,57
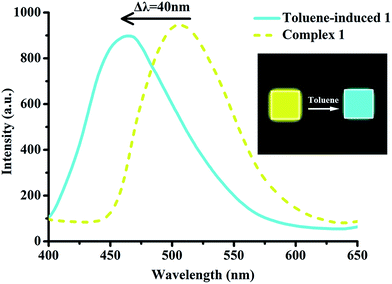 |
| | Fig. 10 Photoluminescent spectra of original and toluene-induced samples of 1. | |
Conclusions
In summary, three novel fluorescent zinc complexes were designed and efficiently synthesized. Their crystal structures, theoretical calculations and photochemical properties were investigated. The complexes exhibit high fluorescent intensity, large Stokes shift, acid–base ON/OFF/ON fluorescence switching properties and low LUMO levels in solid state, which indicates their potential as luminescent materials in devices. Complex 2 exhibited a 3–4 fold higher fluorescence quantum yield (0.45) comparing with complexes 1 and 3. Crystallographic analysis revealed that weaker π–π stacking and less intermolecular interactions were responsible for the superior fluorescence feature of 2. More importantly, complex 1 can serve as a chemosensor to selectively detect toluene with naked eye. This phenomenon may be attributed to the presence of excimer with appropriate substituents and space group. It is important to note that complex 1 is perfectly air-stable and easy to synthesize from readily accessible, inexpensive materials. Those advantages suggest that this new material has a great potential to be utilized as an efficient mean for environment detection etc.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21371135), Tianjin Municipal Natural Science Foundation (No. 13JCZDJC28200).
Notes and references
- K. Li, Y. Chen, W. Lu, N. Zhu and C. M. Che, Chem.–Eur. J., 2011, 17, 4109–4112 CrossRef CAS PubMed.
- Q. Zhao, T. Cao, F. Li, X. Li, H. Jing, T. Yi and C. Huang, Organometallics, 2007, 26, 2077–2081 CrossRef CAS.
- T. Hirano, K. Kikuchi, Y. Urano, T. Higuchi and T. Nagano, J. Am. Chem. Soc., 2000, 122, 12399–12400 CrossRef CAS.
- H. Wang, L. Xue and H. Jiang, Org. Lett., 2011, 13, 3844–3847 CrossRef CAS PubMed.
- H. Sun, H. Guo, W. Wu, X. Liu and J. Zhao, Dalton Trans., 2011, 40, 7834–7841 RSC.
- C. RajáLohani, Analyst, 2010, 135, 2079–2084 RSC.
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536–5537 CrossRef CAS PubMed.
- M. G. Choi, S. Cha, H. Lee, H. L. Jeon and S. K. Chang, Chem. Commun., 2009, 7390–7392 RSC.
- F. Y. Wu, X. F. Tan, Y. M. Wu and Y. Q. Zhao, Spectrochim. Acta, Part A, 2006, 65, 925–929 CrossRef PubMed.
- J. F. Zhang, S. Kim, J. H. Han, S. J. Lee, T. Pradhan, Q. Y. Cao, S. J. Lee, C. Kang and J. S. Kim, Org. Lett., 2011, 13, 5294–5297 CrossRef CAS PubMed.
- S. H. Li, W. T. Yuan, C. Q. Zhu and J. G. Xu, Anal. Biochem., 2004, 331, 235–242 CrossRef CAS PubMed.
- M. M. F. Choi and D. Xiao, Anal. Chim. Acta, 2000, 403, 57–65 CrossRef CAS.
- K. Shirai, M. Matsuoka and K. Fukunishi, Dyes Pigm., 2000, 47, 107–115 CrossRef CAS.
- M. L. Ho, F. M. Hwang and P. N. Chen, et al., Org. Biomol. Chem., 2006, 4, 98–103 CAS.
- Y. Zhang, R. Yang, F. Liu and K. A. Li, Anal. Chem., 2004, 76, 7336–7345 CrossRef CAS PubMed.
- A. K. Singh, U. P. Singh, S. Mehtab and V. Aggarwal, Sens. Actuators, B, 2007, 125, 453–461 CrossRef CAS.
- S. Khatua, S. H. Choi, J. Lee, K. Kim, Y. Do and D. G. Churchill, Inorg. Chem., 2009, 48, 2993–2999 CrossRef CAS PubMed.
- M. M. Ardakani, A. Sadeghi and M. Salavati-Niasari, Talanta, 2005, 66, 837–843 CrossRef CAS PubMed.
- P. Wang, Z. Hong, Z. Xie, S. Tong, O. Wong, C. S. Lee, N. Wong, L. Hung and S. Lee, Chem. Commun., 2003, 14, 1664–1665 RSC.
- Z. Zhang, B. Xu, J. Su, L. Shen, Y. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654–11657 CrossRef CAS PubMed.
- Y. M. Chung, B. Raman, D. S. Kim and K. H. Ahn, Chem. Commun., 2006, 186–188 RSC.
- M. Matsui, Y. Ando, O. Tokura, Y. Kubota and K. Funabiki, Tetrahedron, 2013, 69, 3410–3414 CrossRef CAS.
- H. Samoto, Y. Fukui, H. Ukai, S. Okamoto, S. Takada, F. Ohashi, J. Moriguchi, T. Ezaki and M. Ikeda, Int. Arch. Occup. Environ. Health, 2006, 79, 558–567 CrossRef PubMed.
- K. C. Kao, Y. H. Tsai, M. C. Lin, C. C. Huang, T. C. Y. Tsao and Y. C. Chen, J. Toxicol., Clin. Toxicol., 2000, 38, 679–681 CrossRef CAS PubMed.
- C. H. Pierce, R. L. Dills, M. S. Morgan, P. Vicini and D. A. Kalman, Int. Arch. Occup. Environ. Health, 1998, 71, 433–444 CrossRef CAS PubMed.
- H. Chan, Hong Kong Med. J., 2005, 11, 50–53 Search PubMed.
- C. Garavini and P. Seren, Biochem. Exp. Biol., 1979, 15, 341–348 CAS.
- C. J. Gordon, T. E. Samsam, W. M. Oshiro and P. J. Bushnell, Neurotoxicol. Teratol., 2007, 29, 228–235 CrossRef CAS PubMed.
- Y. Miyagi, F. Shima, K. Ishido, T. Yasutake and K. Kamikaseda, J. Neurol., Neurosurg. Psychiatry, 1999, 66, 794–796 CrossRef CAS.
- F. W. Lavoie, M. C. Dolan, D. F. Danzl and R. L. Barber, Ann. Emerg. Med., 1987, 16, 1266–1273 CrossRef CAS PubMed.
- M. Yücel, M. Takagi, M. Walterfang and D. I. Lubman, Neurosci. Biobehav. Rev., 2008, 32, 910–926 CrossRef PubMed.
- T. M. Goodwin, Obstet. Gynecol., 1988, 71, 715–718 CAS.
- M. Soffritti, F. Belpoggi, D. Cevolani, M. Guarino, M. Padovani and C. Maltoni, Ann. N. Y. Acad. Sci., 2002, 982, 46–69 CrossRef CAS PubMed.
- Ó. Ezquerro, G. Ortiz, B. Pons and M. a. T. Tena, J. Chromatogr. A, 2004, 1035, 17–22 CrossRef PubMed.
- N. Gayathri and N. Balasubramanian, Anal. Lett., 2000, 33, 3037–3050 CrossRef CAS.
- Y. Bao, B. Liu, H. Wang, J. Tian and R. Bai, Chem. Commun., 2011, 47, 3957–3959 RSC.
- A. Castiñeiras, I. García-Santos and M. Saa, Z. Anorg. Allg. Chem., 2008, 634, 2281–2290 CrossRef.
- M. del Carmen Aguirre, J. Borrás, A. Castiñeiras, J. M. García-Monteagudo, I. García-Santos, J. Niclós and D. X. West, Eur. J. Inorg. Chem., 2006, 2006, 1231–1244 CrossRef.
- J. Pflugrath, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1718–1725 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for X-ray Crystal Structure Solution, Göttingen University, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- W. Liu, L. Xu, R. Sheng, P. Wang, H. Li and S. Wu, Org. Lett., 2007, 9, 3829–3832 CrossRef CAS PubMed.
- X. Cheng, H. Jia, T. Long, J. Feng, J. Qin and Z. Li, Chem. Commun., 2011, 47, 11978–11980 RSC.
- Z. Li, M. Yu, L. Zhang, M. Yu, J. Liu, L. Wei and H. Zhang, Chem. Commun., 2010, 46, 7169–7171 RSC.
- J. S. Wu, W. M. Liu, X. Q. Zhuang, F. Wang, P. F. Wang, S. L. Tao, X. H. Zhang, S. K. Wu and S. T. Lee, Org. Lett., 2007, 9, 33–36 CrossRef CAS PubMed.
- H. S. Jung, K. C. Ko, J. H. Lee, S. H. Kim, S. Bhuniya, J. Y. Lee, Y. Kim, S. J. Kim and J. S. Kim, Inorg. Chem., 2010, 49, 8552–8557 CrossRef CAS PubMed.
- A. Patra, S. P. Anthony and T. Radhakrishnan, Adv. Funct. Mater., 2007, 17, 2077–2084 CrossRef CAS.
- Y. Kubota, H. Hara, S. Tanaka, K. Funabiki and M. Matsui, Org. Lett., 2011, 13, 6544–6547 CrossRef CAS PubMed.
- H. C. Yeh, W. C. Wu, Y. S. Wen, D. C. Dai, J. K. Wang and C. T. Chen, J. Org. Chem., 2004, 69, 6455–6462 CrossRef CAS PubMed.
- M. Shimizu, Y. Takeda, M. Higashi and T. Hiyama, Angew. Chem., 2009, 121, 3707–3710 CrossRef.
- S. Mukherjee and P. Thilagar, Phys. Chem. Chem. Phys., 2014, 16, 20866–20877 RSC.
- Y. Ooyama, S. Yoshikawa, S. Watanabe and K. Yoshida, Org. Biomol. Chem., 2007, 5, 1260–1269 CAS.
- Y. Zhou, Y. Xiao, D. Li, M. Fu and X. Qian, J. Org. Chem., 2008, 73, 1571–1574 CrossRef CAS PubMed.
- X. Cheng, Z. Zhang, H. Zhang, S. Han, K. Ye, L. Wang, H. Zhang and Y. Wang, J. Mater. Chem. C, 2014, 2, 7385–7391 RSC.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- Z. Fei, N. Kocher, C. J. Mohrschladt, H. Ihmels and D. Stalke, Angew. Chem., Int. Ed., 2003, 42, 783–787 CrossRef CAS PubMed.
- J. Pang, E. J. P. Marcotte, C. Seward, R. S. Brown and S. Wang, Angew. Chem., 2001, 113, 4166–4169 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Further spectroscopic data like IR, NMR and UV/vis as well as crystallographic tables. CCDC 1433220, 1433094 and 1433095. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra06412d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.