
The effect of Ce on a high-efficiency CeO2/WO3–TiO2 catalyst for the selective catalytic reduction of NOx with NH3
Yang Genga,
Haili Huanga,
Xiaoling Chena,
Hongyu Dinga,
Shijian Yanga,
Fudong Liu†
b and
Wenpo Shan*a
aJiangsu Key Laboratory of Chemical Pollution Control and Resources Reuse, School of Environmental and Biological Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: wenposhan@hotmail.com; Fax: +86 25 84315173; Tel: +86 18012920637
bMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley 94720, California, USA
First published on 4th July 2016
Abstract
In this study, W0.1TiOx and CeaW0.1TiOx (a = 0.1, 0.2, 0.5, 1.0) catalysts were prepared by a novel stepwise precipitation approach. The CeaW0.1TiOx catalysts showed much better activities and N2 selectivity than W0.1TiOx. Particularly, the Ce0.2W0.1TiOx (CeO2/WO3–TiO2) catalyst showed excellent catalytic performance in a broad temperature range from 200 to 450 °C, under a high GHSV condition of 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. Characterizations revealed that the novel preparation approach can achieve highly dispersed Ce species on the surface of the catalyst, and the Ce species could induce enhanced charge imbalance, superior redox functions, and outstanding adsorption and activation properties for NOx and NH3, which is the main reason for the highly efficient NOx abatement capability of the CeO2/WO3–TiO2 catalyst.
000 h−1. Characterizations revealed that the novel preparation approach can achieve highly dispersed Ce species on the surface of the catalyst, and the Ce species could induce enhanced charge imbalance, superior redox functions, and outstanding adsorption and activation properties for NOx and NH3, which is the main reason for the highly efficient NOx abatement capability of the CeO2/WO3–TiO2 catalyst.
Introduction
Selective catalytic reduction of NOx with NH3, using vanadium-based catalysts, has been widely applied for the removal of NOx generated from stationary sources since the 1970s.1 This technology has also been introduced into the market of diesel vehicles and has become the dominant technology for meeting the ever tightened emission standards.2,3 Vanadium-based catalysts, especially V2O5–WO3/TiO2, were used as the first generation of NH3-SCR catalysts for diesel engines.2,4,5 However, the toxicity of the active vanadium species and the narrow operation temperature window are problems for their applications in diesel vehicles.Increasingly stringent emission legislations for NOx in mobile applications will require the use of intensification of NOx reduction aftertreatment technology, which requires that the SCR catalyst should work efficiently under high space velocity conditions.4 For the purpose of obtaining higher deNOx efficiency, great efforts have recently been focused on the optimization of the SCR catalysts for broadening the active deNOx temperature window as widely as possible.2
Many types of vanadium-free catalysts, including oxides and zeolites, based on transition metals and/or rare earth metals have been studied for the NH3-SCR reaction.6,7 Several transition metals such as Fe,8 Mn,9 and Cu10 have been used in NH3-SCR catalysts, while the investigation of rare earth metals for NH3-SCR were mainly focused on Ce.11 Ce has been shown to be an outstanding component in the support of NH3-SCR catalyst.12 Ce is also a very effective promoter for NH3-SCR catalysts, such as V2O5–WO3/TiO2,13 Fe-ZSM-5,14 and Mn based low temperature SCR catalysts.15–17 In addition, Ce-based composite oxide catalysts, such as Ce–Ti,18 Ce–W,19 Ce–Mo,20 Ce–W–Ti,21 and Ce–Cu–Ti22 oxides, were shown to be very effective for NH3-SCR reaction. In our previous study, a high-efficient Ce–W–Ti oxide catalyst was prepared by a novel stepwise precipitation approach.23 This catalyst showed excellent catalytic performance, with superior low-temperature NOx conversion, high N2 selectivity and broad operation temperature window. Even under an extremely high space velocity condition of 1000000 h−1, the catalyst could still be highly effective for NOx conversion to N2. In this study, we furtherly characterized this catalyst using various techniques and investigated the effects of Ce species on the catalyst for NH3-SCR.
Experimental
Catalyst synthesis and catalytic performance test
The CeaW0.1TiOx (“a” represents the Ce/Ti molar ratio; a = 0.1, 0.2, 0.5, 1.0), with a fixed W/Ti molar ratio of 0.1, were prepared by an optimized precipitation method. Ce(NO3)3·6H2O, (NH4)10W12O41 and Ti(SO4)2 were dissolved into distilled water together, with specific Ce/W/Ti molar ratio, and excessive urea was added into the mixed solution. Then, the solution was heated to 90 °C and hold there for 12 h under vigorous stirring. After that, the precipitated solid was collected by filtration, washed with distilled water, dried at 100 °C for 12 h, and calcinated at 500 °C for 5 h, orderly. The Ce0.2W0.1TiOx catalyst with the best NH3-SCR performance was denoted as CeO2/WO3–TiO2. In addition, a W0.1TiOx catalyst was also prepared using the same method as a reference sample.Before NH3-SCR performance test, the powder samples were pressed, crushed and sieved to 40–60 mesh. The SCR performance tests of the sieved powder catalysts (0.06 mL, about 0.07 g) were carried out in a fixed-bed quartz flow reactor at atmospheric pressure. The performance test in this study was carried out under simulated conditions, which is not directly adapted to the NH3-SCR process in realistic conditions. The reaction conditions were controlled as follows: 500 ppm NO, 500 ppm NH3, 5 vol% O2, N2 balance, and 400 mL min−1 total flow rate. The effluent gas, including NO, NH3, NO2 and N2O, was continuously analyzed by an online FTIR gas analyzer (Nicolet Antaris IGS analyzer) equipped with a gas cell of 200 mL volume and 2 m path length and a liquid nitrogen cooled MCT-A detector. The concentration data were collected after 0.5 h when the SCR reaction reached a steady state.
Characterizations
The surface areas of the catalysts were obtained from N2 adsorption/desorption analysis at −196 °C using a Micromeritics ASAP 2020. Prior to N2 physisorption, the catalysts were degassed at 300 °C for 4 h. The surface areas were determined by BET equation in 0.05–0.35 partial pressure range.Powder X-ray diffraction (XRD) measurements of the samples were carried out on a computerized Bruker-AXS D8 diffractometer with Cu Kα (λ = 0.15406 nm) radiation. The data of 2θ were collected at 8° min−1 from 20 to 80°.
The XPS data were obtained on a Scanning X-ray Microprobe (ESCALAB 250, Thermo-VG Scientific) using Al Ka radiation (1486.7 eV). Binding energies of Ce 3d, W 4f, Ti 2p and O 1s were calibrated using C 1s peak (BE = 284.8 eV) as standard.
The surface morphology and elemental composition of the samples were studied using a scanning electron microscope (SEM, FEI Quanta 250F) combined with an energy dispersive X-ray (EDX) attachment. The accelerating voltage was 9.0 kV.
The H2-TPR tests were carried out on a Micromeritics AutoChem_II_2920 chemisorption analyzer. The samples (100 mg) in a quartz reactor were pretreated at 400 °C in a flow of air (50 mL min−1) for 1 h and cooled down to room temperature. Then H2-TPR was performed in 10 vol% H2/Ar gas flow of 50 mL min−1 at a heating rate of 10 °C min−1.
The NOx-TPD and NH3-TPD were performed using the same reaction system as catalytic performance tests. A typical experiment of NOx-TPD used 300 mg sample and a gas flow rate of 200 mL min−1. The experiment consisted of four stages: (1) degasification of the catalyst under N2 at 350 °C for 1 h, (2) adsorption of 500 ppm NO and 5 vol% O2 at 50 °C for 1 h, (3) isothermal desorption under N2 at 50 °C for 1 h, and (4) temperature-programmed desorption under N2 at 10 °C min−1 up to 500 °C. A typical experiment of NH3-TPD used 100 mg sample and a gas flow rate of 200 mL min−1. The experiment procedure of NH3-TPD was similar with that of NOx-TPD, but in stage (2) the adsorption was carried out under 500 ppm NH3 condition.
Results and discussion
NH3-SCR performance
Fig. 1(A) shows the NOx conversion over the catalysts with different Ce contents under a high GHSV of 400000 h−1. The W0.1TiOx just exhibited good catalytic performance in the high temperature region above 400 °C. When Ce was added, all of the CeaW0.1TiOx catalysts showed much better activities. Particularly, the Ce0.2W0.1TiOx catalyst showed excellent catalytic performance in a broad temperature range from 200 to 450 °C, under such a high space velocity condition.
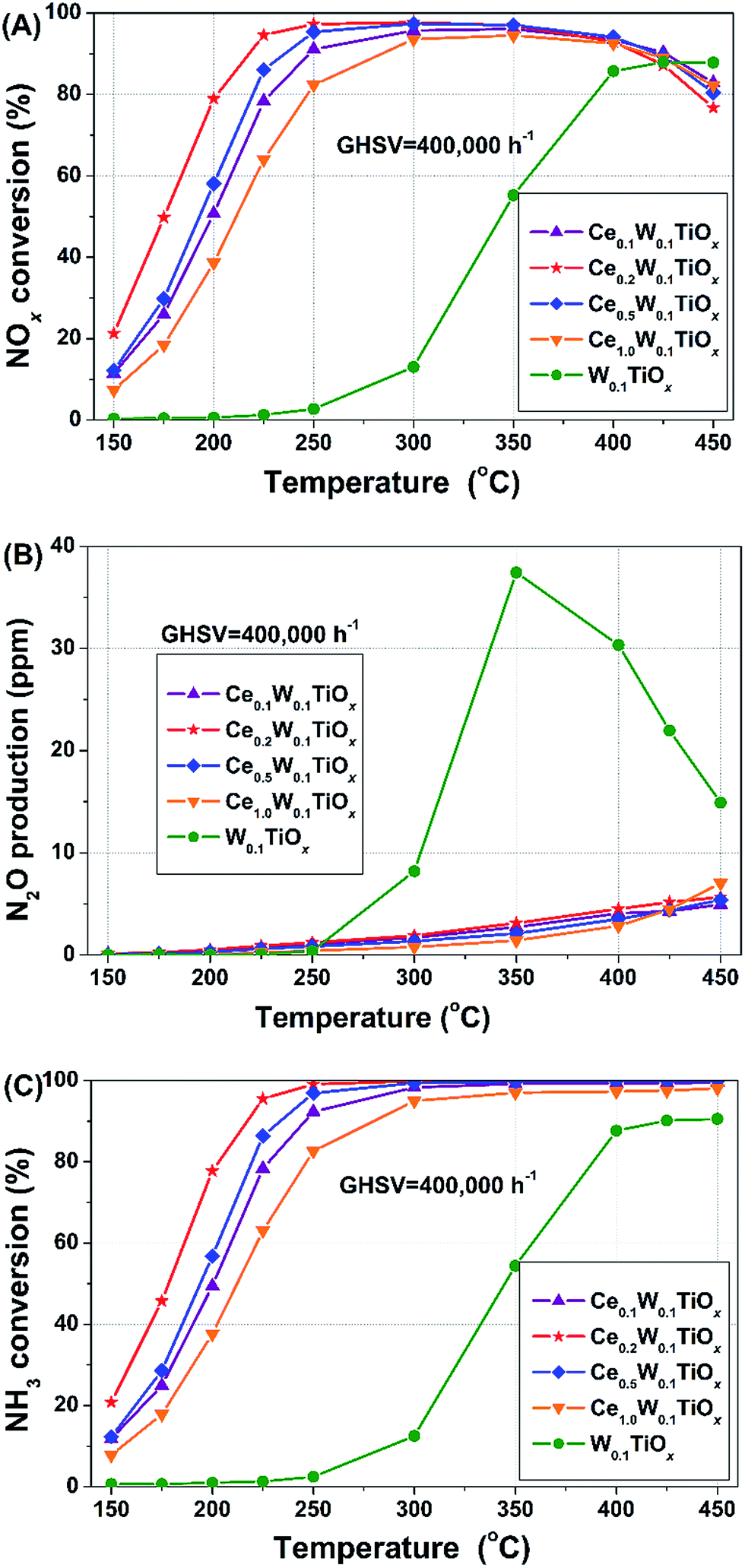 | ||
| Fig. 1 (A) NOx conversions, (B) N2O productions and (C) NH3 conversions over the W0.1TiOx and CeaW0.1TiOx catalysts. Reaction conditions: [NO] = [NH3] = 500 ppm, [O2] = 5 vol%, N2 balance and GHSV = 400000 h−1. | ||
The N2O production over the samples during the catalytic performance test is shown in Fig. 1(B). We can see that, the addition of Ce remarkably inhibited the formation of N2O during NH3-SCR. At 350 °C, the N2O production over W0.1TiOx reached 37.4 ppm. When a small amount of Ce was added, the N2O production over Ce0.1W0.1TiOx dropped to be just 2.7 ppm. The peak value of N2O over W0.1TiOx at 350 °C was resulted from the competition between non-selective catalytic reduction reaction (NSCR reaction, 4NO + 4NH3 + 3O2 → 4N2O + 6H2O) and SCR reaction (4NO + 4NH3 + O2 → 4N2 + 6H2O).24,25
The NH3 conversions over the samples during the tests are shown in Fig. 1(C). Below 350 °C, the NH3 conversion over each CeaW0.1TiOx catalyst was very similar with the corresponding NOx conversion, indicating that the NO and NH3 were almost consumed with the same molar ratio. Above 350 °C, all of the NH3 conversions over CeaW0.1TiOx catalysts kept to be 100%, which is associated with the oxidation of NH3 in addition to SCR reaction. The NH3 conversion over W0.1TiOx at 350 °C was almost the same as the NO conversion, indicating that the two N atoms in N2O were mainly due to the coupling of one nitrogen atom from NH3 and another one from NO.26 A comparative investigation of the N2O formation mechanisms during NH3-SCR over Ce0.2W0.1TiOx and W0.1TiOx demonstrated that CeO2 is very effective for enhancing SCR reaction rate and thus depressing the NSCR reaction due to the competition between these two reactions.24
NO oxidation performance
To investigate the NO oxidation properties of the catalysts, separated NO oxidation tests were carried out over the samples. NO2 productions during the separated NO oxidations are shown in Fig. 2. NO oxidation to NO2 is very important for low-temperature SCR to promote deNOx efficiency by accelerating the “fast SCR” process (2NH3 + NO + NO2 → 2N2 + 3H2O). If a catalyst can effectively oxidize NO to NO2 in situ under NH3-SCR conditions, the NOx can be efficiently removed at low temperature.27–29 In this study, the order of NO2 production over the catalysts matched perfectly with that of the low-temperature NOx conversion. The NO2 productions over CeaW0.1TiOx catalysts were clearly higher than that over W0.1TiOx, implying that the addition of Ce could effectively promote the oxidation of NO during NH3-SCR reaction and thereby enhanced the low-temperature SCR performance.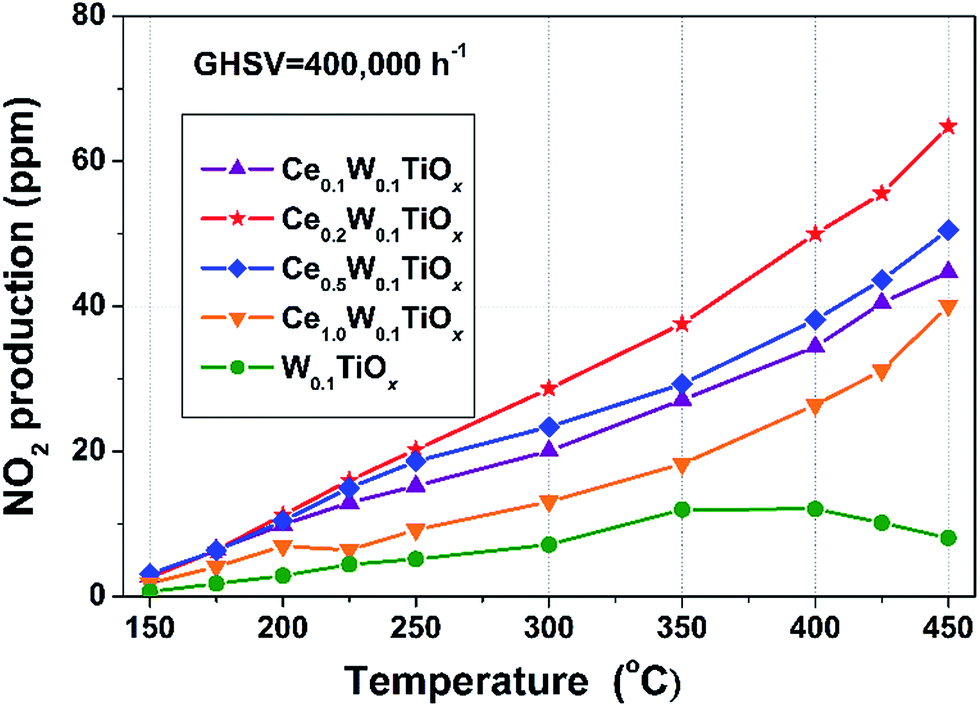 | ||
| Fig. 2 NO2 production during separate NO oxidation reaction over W0.1TiOx and CeaW0.1TiOx catalysts. Reaction conditions: [NO] = 500 ppm, [O2] = 5 vol%, N2 balance and GHSV = 400000 h−1. | ||
BET surface area and XRD
As shown in Table 1, the BET surface area of W0.1TiOx, Ce0.1W0.1TiOx and Ce0.2W0.1TiOx were similar with each other. With further increase of Ce, the BET surface area decreased significantly from Ce0.2W0.1TiOx to Ce0.5W0.1TiOx and sharply from Ce0.5W0.1TiOx to Ce1.0W0.1TiOx. Among these samples, the BET surface area of Ce0.2W0.1TiOx was the highest one, which probably be a reason for the highest NH3-SCR performance of Ce0.2W0.1TiOx.| Sample | BET surface area (m2 g−1) | NOx desorption (μmol g−1) | NH3 desorption (μmol g−1) |
|---|---|---|---|
| W0.1TiOx | 167.2 | 5.6 | 487.6 |
| Ce0.1W0.1TiOx | 160.5 | 14.0 | 419.7 |
| Ce0.2W0.1TiOx | 173.6 | 24.2 | 449.0 |
| Ce0.5W0.1TiOx | 127.5 | 6.7 | 391.9 |
| Ce1.0W0.1TiOx | 8.9 | 7.7 | 366.2 |
The XRD results of the samples are shown in Fig. 3. Only anatase TiO2 was detected in W0.1TiOx. With the addition of Ce, the crystallization of anatase TiO2 got weaker and weaker, but still no Ce or W species was observed in Ce0.1W0.1TiOx or Ce0.2W0.1TiOx. With further increase of Ce, clear cubic CeO2 was observed in Ce0.5W0.1TiOx and Ce1.0W0.1TiOx and Ce2WO6 was observed in Ce1.0W0.1TiOx. The coexistence of Ce with W and Ti species in Ce0.2W0.1TiOx could significantly inhibit the crystallization of Ti species, and the Ce and W species were probably existed as amorphous phase or crystallite phase with very small particle size, which will lead to highly dispersed active species.30 In such case, it is reasonable to see high efficient NOx conversion over Ce0.2W0.1TiOx together with no clear XRD diffraction peak of Ce or W species.
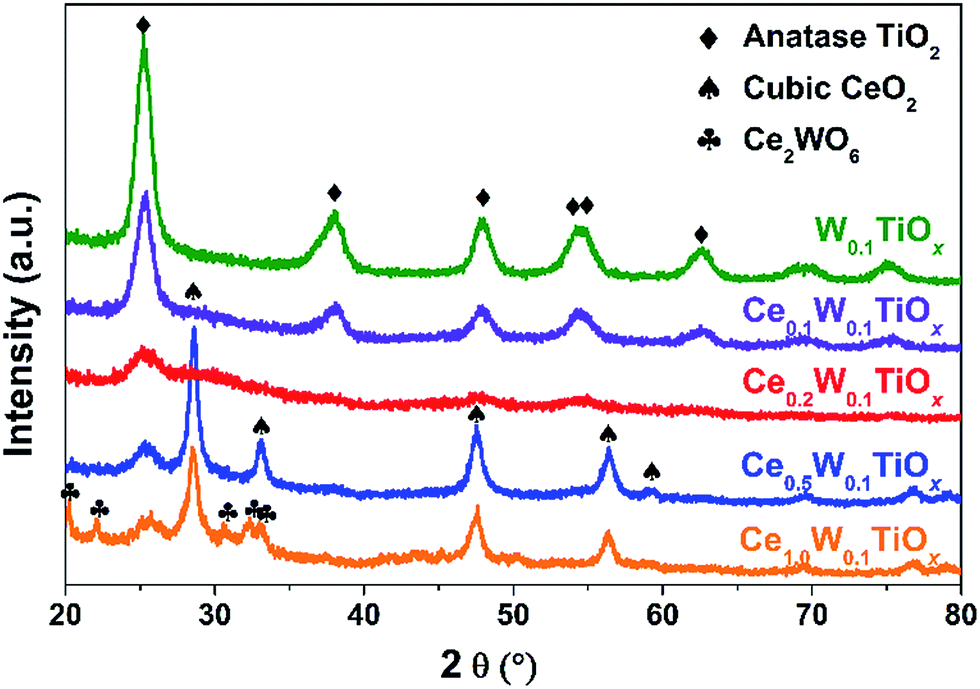 | ||
| Fig. 3 XRD patterns of the W0.1TiOx and CeaW0.1TiOx catalysts. | ||
Overall structure of the catalyst
Fig. 4 shows the SEM images of the W0.1TiOx and CeaW0.1TiOx catalysts. All the samples consisted of small spherical particles with diameters in micrometer level. The particle size of these samples observed with SEM was much larger than the average crystallite sizes of TiO2 calculated by XRD (in nanometer level), indicating that each spherical particle was not a single crystallite but an agglomerate of many single crystallites. | ||
| Fig. 4 SEM images of the W0.1TiOx and CeaW0.1TiOx catalysts. | ||
To investigate the atomic distributions of the metal elements, two techniques (SEM-EDX and XPS) of elemental composition analysis were carried out. The resolution of EDX is over 1 μm in depth, so its result were used to represent the overall status of the sample. For XPS, the detection depth is less than 10 nm, so its result can be used as the surface information of the sample. The analysis results by EDX and XPS are shown in Fig. 5. From the EDX results, we can see that the measured Ce/Ti molar ratios were similar with the added molar ratios. However, the surface Ce/Ti molar ratios for Ce0.2W0.1TiOx, Ce0.5W0.1TiOx and Ce1.0W0.1TiOx measured by XPS were much higher than the corresponding added molar ratios. The comparison of surface and overall Ce/Ti molar ratios clearly indicated the enrichment of Ce on the surface of the catalyst, which demonstrated the structure of the CeO2/WO3–TiO2 catalyst with dispersion of CeO2 on WO3–TiO2. An EDX mapping of the Ce for the Ce0.2W0.1TiOx catalyst in Fig. 6 showed that Ce was distributed evenly.
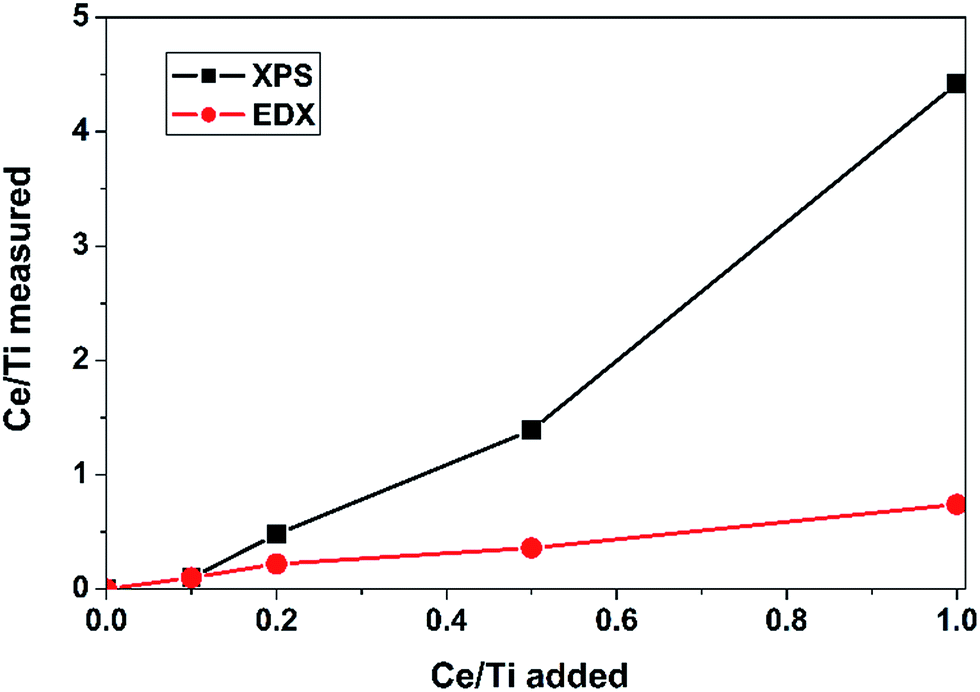 | ||
| Fig. 5 The measured Ce/Ti molar ratio of the W0.1TiOx and CeaW0.1TiOx catalysts by SEM-EDX and XPS. | ||
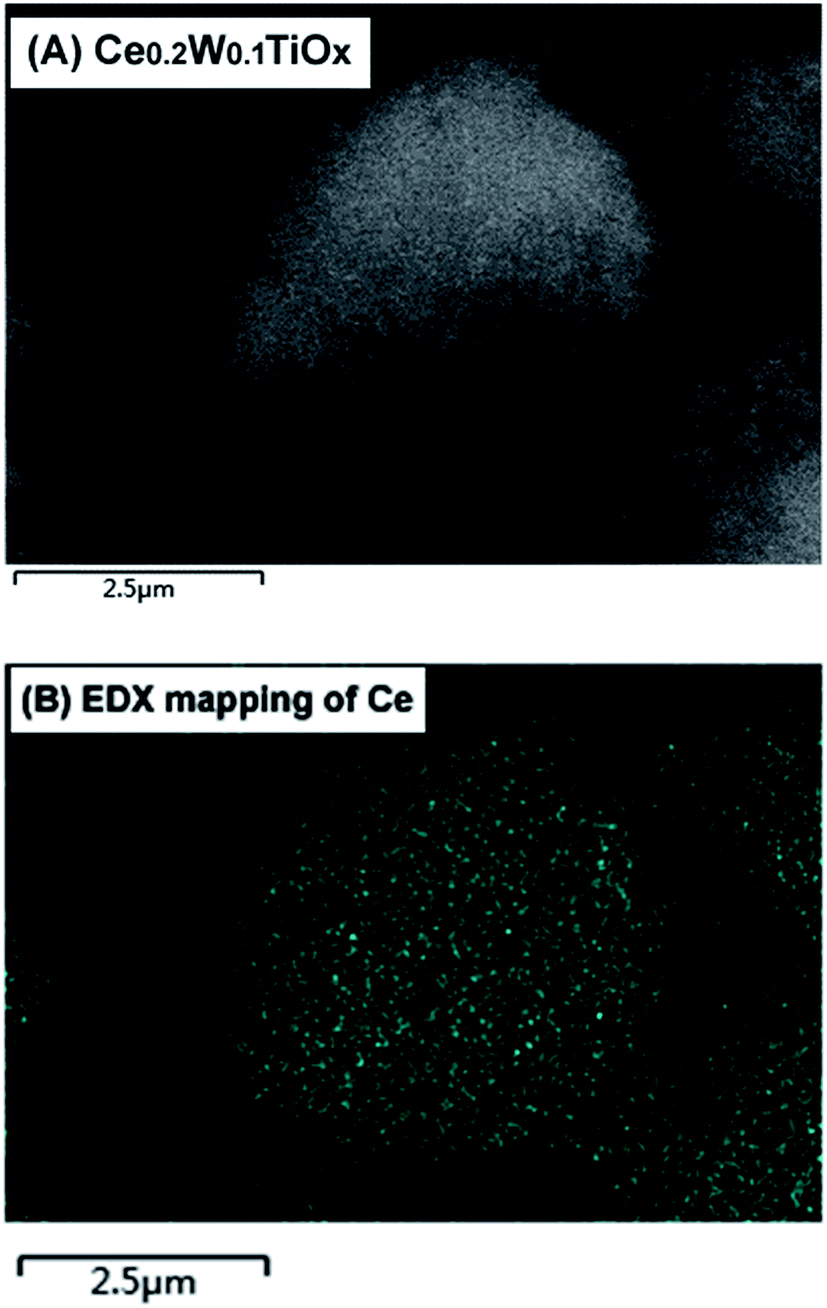 | ||
| Fig. 6 EDX mapping of the Ce for the Ce0.2W0.1TiOx catalyst. | ||
NOx/NH3-TPD
To investigate the NOx and NH3 adsorption/desorption capabilities of the catalysts, NOx-TPD and NH3-TPD were performed on CeaW0.1TiOx and W0.1TiOx (Fig. 7).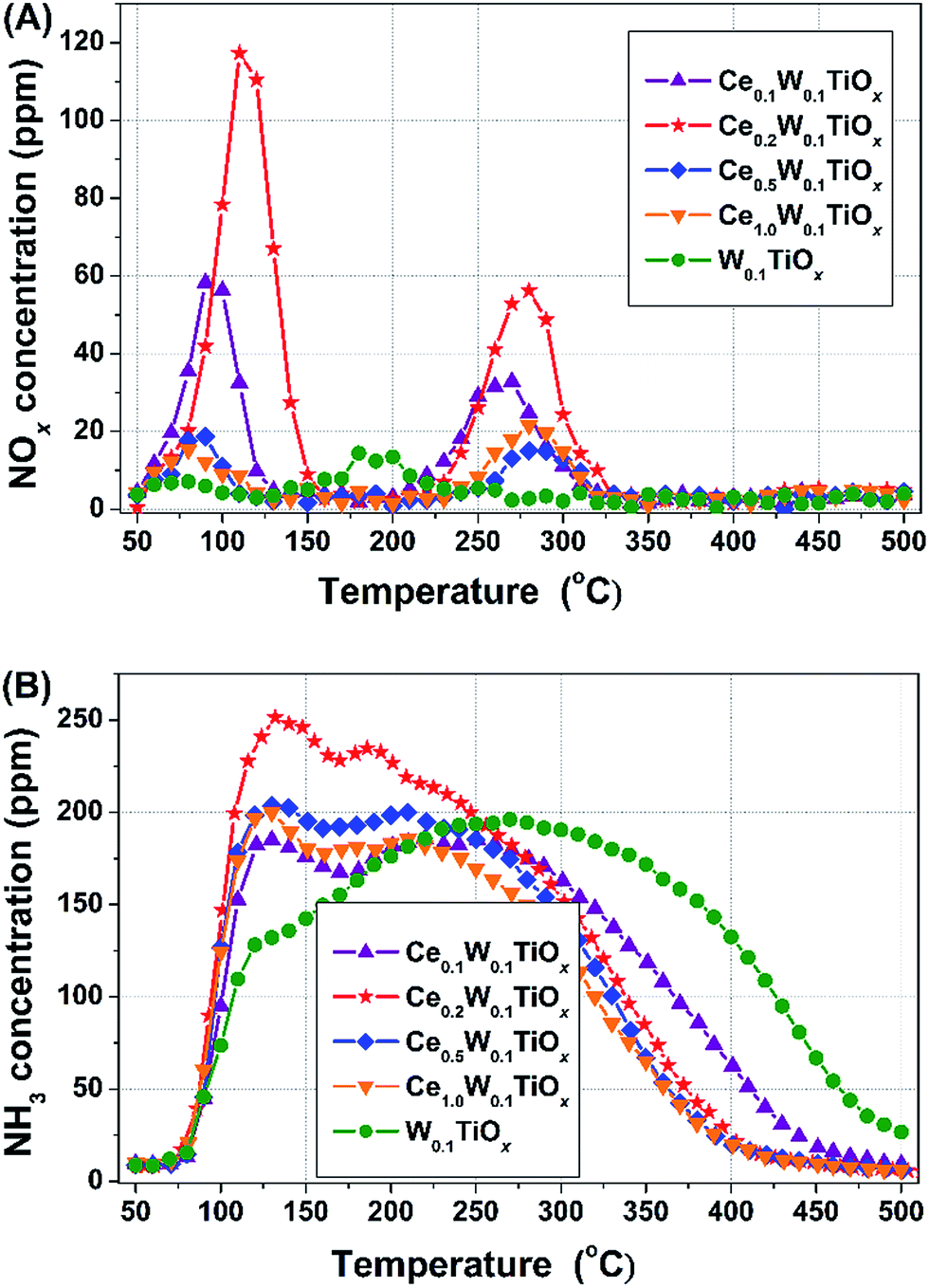 | ||
| Fig. 7 (A) NOx-TPD and (B) NH3-TPD profiles of the W0.1TiOx and CeaW0.1TiOx catalysts. | ||
The NOx-TPD profiles are shown in Fig. 7(A). The first NOx peak of CeaW0.1TiOx at ca. 100 °C was due to the desorption of physisorbed NOx, while the second NOx peak at ca. 275 °C was mainly associated with the decomposition of chemsorbed NOx species.31,32 The two NOx peaks of W0.1TiOx were both observed at lower temperature than those of CeaW0.1TiOx, indicating that the adsorption of NOx on W0.1TiOx was much weaker than those on CeaW0.1TiOx. The adsorbed NOx on W0.1TiOx was obviously less than that on Ce0.1W0.1TiOx or Ce0.2W0.1TiOx, with similar BET surface area, indicating that NOx was mainly adsorbed on Ce sites. The adsorbed NOx on Ce0.5W0.1TiOx and Ce1.0W0.1TiOx were clearly less than that on Ce0.1W0.1TiOx or Ce0.2W0.1TiOx, indicating that the accumulation of Ce on the surface of catalysts with high Ce contents (as detected by XRD) was unfavourable for NOx adsorption. Therefore, highly dispersed Ce species on the Ce0.2W0.1TiOx catalyst is very important for the adsorption and activation of NOx.
It has been indicated that surface acidity plays an important role in the adsorption and activation of NH3, and the SCR reaction in the high-temperature region is likely controlled by the surface acid properties.33,34 Both of W and Ti can play as acid sites for NH3 adsorption during NH3-SCR reaction. Therefore, it is reasonable to see that the addition of Ce induced less NH3 adsorption on CeaW0.1TiOx than that on W0.1TiOx (Fig. 7(B) and Table 1). Furthermore, the addition of Ce led to a shift of NH3 desorption peak to the low temperature from W0.1TiOx to CeaW0.1TiOx. It indicates that the NH3 adsorbed species could be more easily desorbed at low temperature with the addition of Ce. Considering BET surface area for Ce1W0.1TiOx is just 8.9 m2 g−1, the amount of adsorbed NH3 (366.2 μmol g−1) is more than that for monolayer adsorption (considering ≈1019 sites per m2 for a monolayer), indicating the existence of physisorption. In addition, NH3 could be chemically adsorbed on both of the Lewis and Brønsted sites to form NH3ads and NH4+ species, which could both present in the NH3-SCR reaction.35–37 Ce0.2W0.1TiOx exhibited much better NH3 adsorption capacity than other CeaW0.1TiOx catalysts (Table 1). That was probably an important reason for the better NH3-SCR performance of Ce0.2W0.1TiOx.
H2-TPR
The H2-TPR profiles of W0.1TiOx and Ce0.2W0.1TiOx are shown in Fig. 8. The three peaks at 535/544, 562/557, and 782/794 °C were associated with the multi-stage reduction process from WO3 to WO2 via two non-stoichiometric WOx oxides.38,39 In addition, two more TPR peaks appeared at 473 and 880 °C, respectively, on Ce0.2W0.1TiOx. The peaks at 473 °C can be attributed to the reduction of surface Ce4+ to Ce3+, while the peak at 880 °C might be assigned to the reduction of bulk CeO2.13,40 The H2-TPR profiles strongly suggest an enhancement of redox property by the Ce species on the catalyst.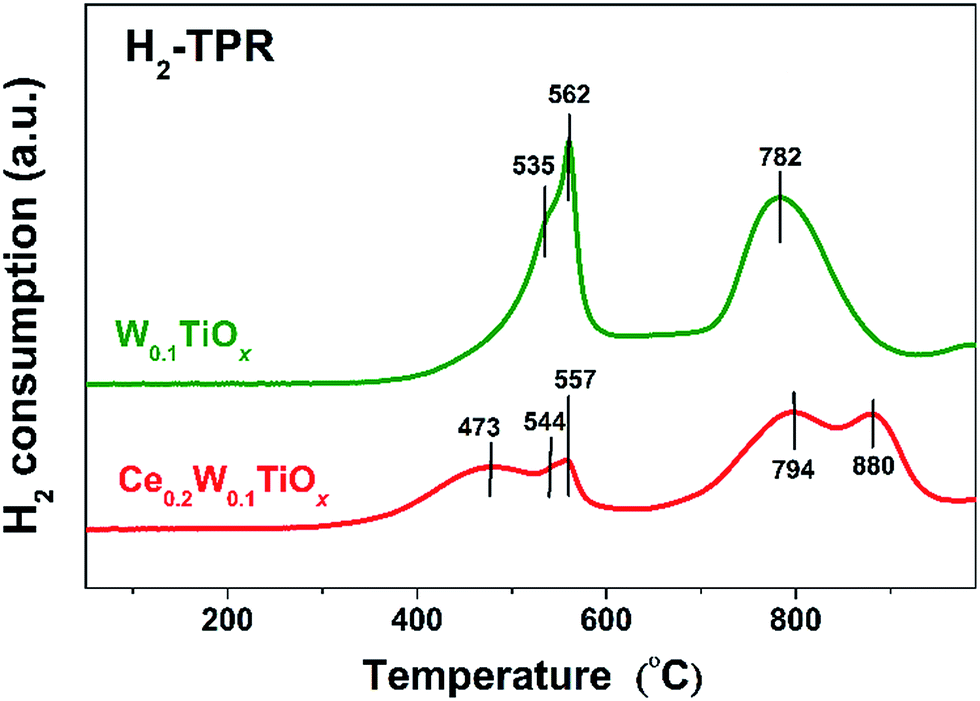 | ||
| Fig. 8 H2-TPR profiles of the W0.1TiOx and Ce0.2W0.1TiOx. | ||
The redox property of catalyst is very important for NH3-SCR. Topsøe et al. proposed a well-known catalytic cycle for the SCR reaction involving both acid-based and redox functions.41 Previous studies by Lietti et al. have demonstrated that the redox functions of NH3-SCR catalyst govern the catalytic reactivity in the low-temperature region.34,42 Therefore, the enhanced redox property of Ce0.2W0.1TiOx due to Ce species would beneficial for the low temperature NH3-SCR performance.
XPS results of O 1s
With the introduction of Ce, the presence of Ce3+ in the catalyst could induce a charge imbalance, which would lead to oxygen vacancies and unsaturated chemical bonds and generate additional chemisorbed oxygen or weakly adsorbed oxygen species on the surface of catalyst.22,43,44The XPS results of O 1s of W0.1TiOx and Ce0.2W0.1TiOx are shown in Fig. 9. The peak of O 1s was fitted into three sub-bands by searching for the optimum combination of Gaussian bands with correlation coefficients (r2) above 0.99. The sub-bands at 529.9–530.4 eV could be attributed to the lattice oxygen O2− (denoted as Oβ). Two shoulder sub-bands at 531.4–531.6 eV and 532.9–533.0 eV are assigned to the surface adsorbed oxygen (denoted as Oα), such as the O22− and O− belonging to defect-oxide or hydroxyl-like group, and chemisorbed water (denoted as O′α), respectively.45,46 The Oα ratio of Ce0.2W0.1TiOx (35.4%) calculated by Oα/(Oα + O′α + Oβ) was much higher than that of W0.1TiOx (12.3%), which means that the introduction of Ce indeed created more surface oxygen vacancies. Oα is usually more reactive than Oβ for oxidation reactions due to its higher mobility.16 Therefore, the enhanced Oα of Ce0.2W0.1TiOx would beneficial for the activation of NO and NH3 in the SCR reaction, and thereby promote the NOx conversion at low temperature.
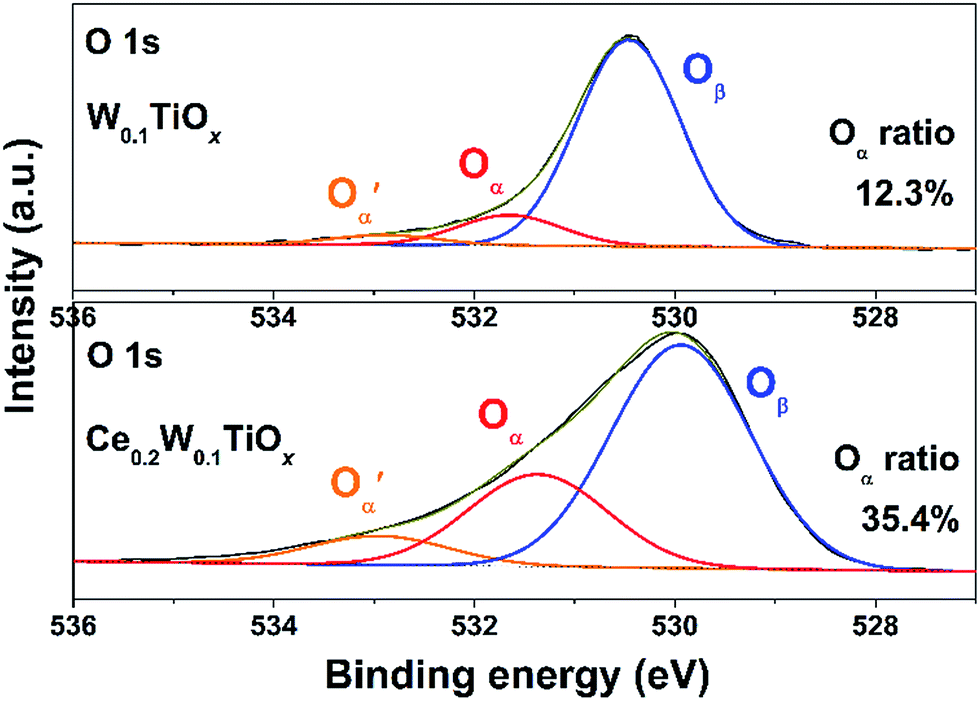 | ||
| Fig. 9 XPS results of O 1s of the W0.1TiOx and Ce0.2W0.1TiOx. | ||
O2 shut off test
Oxygen plays an important role in the NH3-SCR reaction.1 To investigate the effect of the lattice oxygen of the catalyst, the NOx conversion of the W0.1TiOx and Ce0.2W0.1TiOx as a function of time was measured at 350 °C when the supply of oxygen was stopped during the NH3-SCR reaction (Fig. 10).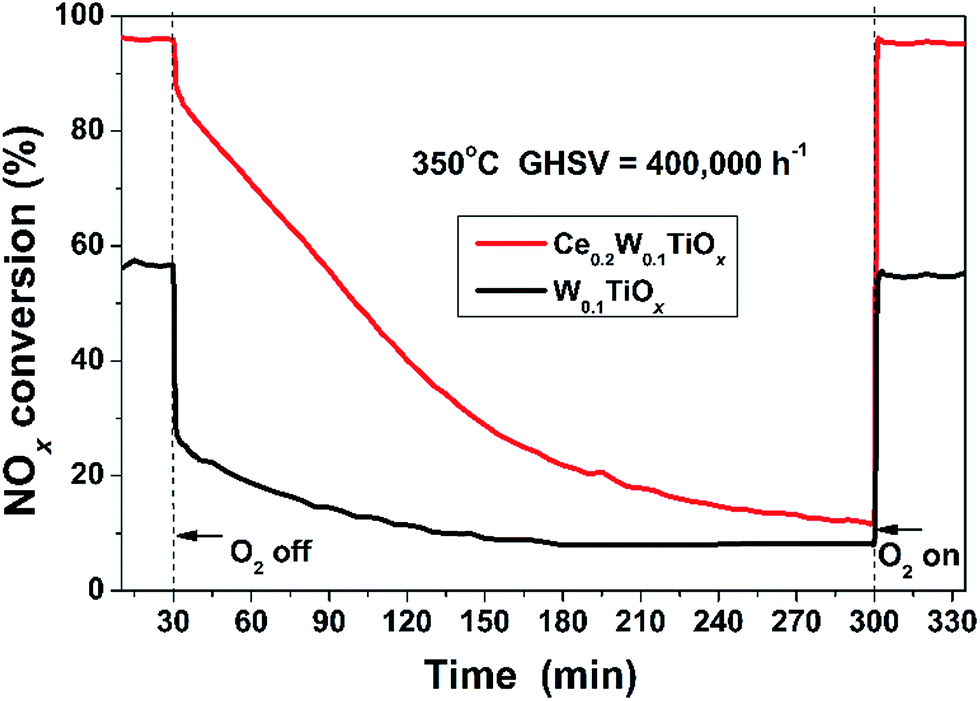 | ||
| Fig. 10 O2 shut off test results of the W0.1TiOx and Ce0.2W0.1TiOx at 350 °C. | ||
Before the supply of oxygen was stopped, the NOx conversion over Ce0.2W0.1TiOx was kept at ca. 96% under the reaction conditions (GHSV = 400000 h−1 and temperature = 350 °C). After the supply of oxygen was stopped, the NOx conversion rapidly decreased by ca. 10% in 2 min, and then the decrease rate changed to be steady. After the stop of O2 for 270 min, the NOx conversion gradually decreased to be ca. 12%. Upon resupply of gaseous oxygen, the NOx conversions of Ce0.2W0.1TiOx was recovered to the initial level immediately. This result demonstrated that the lattice oxygen of the catalyst has substantially participated in the NH3-SCR reaction in the absence of gaseous oxygen.47 Under the same reaction condition, the initial NOx conversion over W0.1TiOx was kept at ca. 57%. As soon as the supply of oxygen was stopped, the NOx conversion decreased sharply to be ca. 26%. After the stop of O2 for 180 min, the NOx conversion kept stably to be ca. 8%. Upon resupply of gaseous oxygen, the NOx conversions of W0.1TiOx was also recovered immediately. The results of O2 shut off experiments showed that the W0.1TiOx only contained small amount of lattice oxygen that could be reduced, and lattice oxygen just slightly affect the reaction. However, the introduction of Ce remarkably increased the amount of lattice oxygen that could be reduced, and enhanced lattice oxygen could participate significantly in the SCR reaction.
Conclusions
W0.1TiOx and CeaW0.1TiOx (a = 0.1, 0.2, 0.5, 1.0) catalysts were prepared by a novel stepwise precipitation approach. The catalysts were characterized by various techniques and the effects of Ce on the catalyst for NH3-SCR were investigated.The W0.1TiOx just exhibited good catalytic performance in the high temperature region above 400 °C. When Ce was added, all of the CeaW0.1TiOx catalysts showed remarkably improved catalytic performance and N2 selectivity. Particularly, the Ce0.2W0.1TiOx (CeO2/WO3–TiO2) catalyst showed excellent catalytic performance in a broad temperature range from 200 to 450 °C, under a high GHSV condition of 400000 h−1.
Characterizations indicated that the CeO2/WO3–TiO2 catalyst prepared by the stepwise precipitation approach can achieve highly dispersed active CeO2 on WO3–TiO2 and intense interaction among Ce, W and Ti species. The Ce species on the catalyst could induce enhanced charge imbalance, superior redox functions, and outstanding adsorption and activation properties of the reactants. That is the reason for the excellent NH3-SCR performance of CeO2/WO3–TiO2 catalyst.
Acknowledgements
We gratefully acknowledge the financial supports from the National Natural Science Foundation of China (51308296), the Fundamental Research Funds for the Central Universities (30920140111012) and the Qing Lan Project of Jiangsu Province, China.References
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- B. Guan, R. Zhan, H. Lin and Z. Huang, Appl. Therm. Eng., 2014, 66, 395–414 CrossRef CAS.
- M. Koebel, M. Elsener and M. Kleemann, Catal. Today, 2000, 59, 335–345 CrossRef CAS.
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155–3207 CrossRef CAS PubMed.
- F. Liu, W. Shan, D. Pan, T. Li and H. He, Chin. J. Catal., 2014, 35, 1438–1445 CrossRef CAS.
- W. Shan and H. Song, Catal. Sci. Technol., 2015, 5, 4280–4288 CAS.
- F. Liu, W. Shan, X. Shi, C. Zhang and H. He, Chin. J. Catal., 2011, 32, 1113–1128 CAS.
- F. Liu, H. He and C. Zhang, Chem. Commun., 2008, 2043–2045 RSC.
- M. Casapu, O. Kröcher and M. Elsener, Appl. Catal., B, 2009, 88, 413–419 CrossRef CAS.
- Z. Si, D. Weng, X. Wu, J. Li and G. Li, J. Catal., 2010, 271, 43–51 CrossRef CAS.
- W. Shan, F. Liu, Y. Yu and H. He, Chin. J. Catal., 2014, 35, 1251–1259 CrossRef CAS.
- Y. Li, H. Cheng, D. Li, Y. Qin, Y. Xie and S. Wang, Chem. Commun., 2008, 1470–1472 RSC.
- L. Chen, J. Li and M. Ge, J. Phys. Chem. C, 2009, 113, 21177–21184 CAS.
- R. Q. Long and R. T. Yang, J. Am. Chem. Soc., 1999, 121, 5595–5596 CrossRef CAS.
- Z. Liu, Y. Yi, S. Zhang, T. Zhu, J. Zhu and J. Wang, Catal. Today, 2013, 216, 76–81 CrossRef CAS.
- Z. Wu, R. Jin, Y. Liu and H. Wang, Catal. Commun., 2008, 9, 2217–2220 CrossRef CAS.
- Z. Liu, J. Zhu, J. Li, L. Ma and S. I. Woo, ACS Appl. Mater. Interfaces, 2014, 6, 14500–14508 CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, ChemCatChem, 2011, 3, 1286–1289 CrossRef CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Chem. Commun., 2011, 47, 8046–8048 RSC.
- Y. Peng, R. Qu, X. Zhang and J. Li, Chem. Commun., 2013, 49, 6215–6217 RSC.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Appl. Catal., B, 2012, 115–116, 100–106 CrossRef CAS.
- Z. Liu, Y. Yi, J. Li, S. I. Woo, B. Wang, X. Cao and Z. Li, Chem. Commun., 2013, 49, 7726–7728 RSC.
- H. Huang, W. Shan, S. Yang and J. Zhang, Catal. Sci. Technol., 2014, 4, 3611–3614 CAS.
- Y. Geng, W. Shan, S. Xiong, Y. Liao, S. Yang and F. Liu, Catal. Sci. Technol., 2016, 6, 3149–3155 CAS.
- S. Yang, S. Xiong, Y. Liao, X. Xiao, F. Qi, Y. Peng, Y. Fu, W. Shan and J. Li, Environ. Sci. Technol., 2014, 48, 10354–10362 CrossRef CAS PubMed.
- P. R. Ettireddy, N. Ettireddy, T. Boningari, R. Pardemann and P. G. Smirniotis, J. Catal., 2012, 292, 53–63 CrossRef CAS.
- F. Liu, W. Shan, Z. Lian, L. Xie, W. Yang and H. He, Catal. Sci. Technol., 2013, 3, 2699–2707 CAS.
- J. Li, H. Chang, L. Ma, J. Hao and R. T. Yang, Catal. Today, 2011, 175, 147–156 CrossRef CAS.
- G. Qi and R. T. Yang, Appl. Catal., B, 2003, 44, 217–225 CrossRef CAS.
- Y. Peng, K. Li and J. Li, Appl. Catal., B, 2013, 140–141, 483–492 CrossRef CAS.
- F. Liu and H. He, J. Phys. Chem. C, 2010, 114, 16929–16936 CAS.
- F. Liu, H. He, Y. Ding and C. Zhang, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS.
- L. Lietti, P. Forzatti and F. Bregani, Ind. Eng. Chem. Res., 1996, 35, 3884–3892 CrossRef CAS.
- L. Lietti, I. Nova and P. Forzatti, Top. Catal., 2000, 11–12, 111–122 CrossRef.
- F. Giraud, C. Geantet, N. Guilhaume, S. Loridant, S. Gros, L. Porcheron, M. Kanniche and D. Bianchi, J. Phys. Chem. C, 2015, 119, 15401–15413 CAS.
- F. Giraud, C. Geantet, N. Guilhaume, S. Loridant, S. Gros, L. Porcheron, M. Kanniche and D. Bianchi, J. Phys. Chem. C, 2014, 118, 15677–15692 CAS.
- Y. Geng, W. Shan, S. Xiong, Y. Liao, S. Yang and F. Liu, Catal. Sci. Technol., 2016, 6, 3149–3155 CAS.
- D. G. Barton, S. L. Soled, G. D. Meitzner, G. A. Fuentes and E. Iglesia, J. Catal., 1999, 181, 57–72 CrossRef CAS.
- Z. Ma, D. Weng, X. Wu, Z. Si and B. Wang, Catal. Commun., 2012, 27, 97–100 CrossRef CAS.
- Z. Liu, S. Zhang, J. Li and L. Ma, Appl. Catal., B, 2014, 144, 90–95 CrossRef CAS.
- N.-Y. Topsøe, Science, 1994, 265, 1217–1219 Search PubMed.
- L. Lietti, P. Forzatti and F. Berti, Catal. Lett., 1996, 41, 35–39 CrossRef CAS.
- T. Gu, Y. Liu, X. Weng, H. Wang and Z. Wu, Catal. Commun., 2010, 12, 310–313 CrossRef CAS.
- C. Liu, L. Chen, H. Chang, L. Ma, Y. Peng, H. Arandiyan and J. Li, Catal. Commun., 2013, 40, 145–148 CrossRef CAS.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- J. Fang, X. Bi, D. Si, Z. Jiang and W. Huang, Appl. Surf. Sci., 2007, 253, 8952–8961 CrossRef CAS.
- S. Moon Lee, S. Su Kim and S. Chang Hong, Chem. Eng. Sci., 2012, 79, 177–185 CrossRef.
Footnote |
| † Current address: BASF Corporation, 25 Middlesex Essex Turnpike, Iselin, New Jersey 08830, United States. |
| This journal is © The Royal Society of Chemistry 2016 |