
Tungsten-promoted titania as solid acid for catalytic hydrolysis of waste bottle PET in supercritical CO2†
Wen-Ze Guo,
Hui Lu,
Xue-Kun Li and
Gui-Ping Cao*
UNILAB, State Key Lab of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: gpcao@ecust.edu.cn; Fax: +86-21-6425-3934; Tel: +86-21-6425-3934
First published on 18th April 2016
Abstract
Tungsten-promoted titania solid acid catalysts were synthesized by a hydrothermal method and used in the hydrolysis of waste bottle polyethylene terephthalate (PET) in supercritical CO2. The structure of the catalytically active sites in this system was determined by XRD, Raman spectroscopy, and HR-TEM. The surface acidity and reduction properties were studied by NH3-TPD, titration experiments, and H2-TPR. The results indicated that the tungsten phase existed as surface WOx species, and a direct relationship among the number of nanoclusters consisting of polytungstate species on the surface, the number of Brønsted acid sites, and the catalytic activity was discovered. Partial reduction of WOx species in the presence of the ethylene glycol produced during hydrolysis was also observed, and the polytungstate species were easier to reduce with increased condensation. A mechanism was proposed to describe the hydrolysis in which water molecules and hydronium ions were carried by supercritical CO2 and penetrated the swollen PET matrix, and the hydrolysis occurred preferentially in the amorphous region of the surface and bulk of the PET matrix. The results reported here may help to pave the way for the design of active, reusable tungsten-based solid acid catalysts and highly efficient reaction systems for the polyester hydrolysis.
1 Introduction
Polyethylene terephthalate (PET) is one of the most important semi-crystalline polymers, and in 2014, its production volume had increased to 29 million tons per year globally.1 Owing to its excellent physicochemical properties, such as glass-like transparency and high strength, PET is used widely in fibers, food containers, engineering resins, and especially in soft drink bottles, which account for more than 60% of total consumption.2 PET is manufactured through condensation polymerization of terephthalic acid (TPA) and ethylene glycol (EG), both of which are derived from petroleum. Up to 2014, the production of PET consumed about 180 million tons of petroleum a year.1 Diminishing nonrenewable fossil fuel reserves mean that it is necessary to develop alternative energy sources and sustainable, environmentally benign processes for recycling materials.Depolymerizing waste bottle PET and recycling the monomer units can decrease dependence on petroleum crude oil, and serve as a partial solution to the severe solid waste problem caused by non-biodegradable PET. There are several chemical recycling methods for waste PET with various low molecular weight products obtained, depending on the regents used to cleave the ester groups in the PET chains. For example, dimethyl terephthalate (DMT) and EG can be obtained by methanolysis,3 bis-hydroxyethyl terephthalate (BHET) and EG are produced by glycolysis,4 and valuable organic amines can be produced by aminolysis or ammonolysis.5 Hydrolysis of PET to TPA and EG has also attracted great attention from both academia and industry because of the development of PET synthesis directly from EG and TPA.6 PET hydrolysis can be classified as acid, alkaline, or neutral,7–11 of which acid hydrolysis is the most common method because of its comparatively mild reaction conditions.6
Ravens et al.12 investigated the heterogeneous hydrolysis rate of PET fibers catalyzed by different concentrations of hydrochloric acid at 70 °C. They concluded that hydrolysis occurred predominantly in the amorphous regions of PET and that the reaction rate was principally determined by the solubility of HCl in these regions. Subsequently, there have been many studies of acid hydrolysis of PET. Yoshioka et al.13 investigated the hydrolysis of waste PET powder in 1–10 M sulfuric acid at 150 °C, and the sulfuric acid could be reused by a recovery method such as dialysis. In their further study of the hydrolytic kinetics and mechanism in sulfuric acid, a modified shrinking-core model was developed to describe the reaction. In the model, the effective surface area is proportional to the degree of PET hydrolysis, and it is affected by the formation and growth of pores and cracks on the PET powder.14 This model was also used to explain the hydrolysis of PET in nitric acid.15 Mishra et al.16 used mixed sulfuric and nitric acid to catalyze the hydrolysis of PET powder, and a similar shrinking-core model was proposed to describe the hydrolysis, indicating that the hydrolysis rate and the apparent rate constant were inversely proportional to the PET particle size. The melting point of PET is 245–265 °C; therefore, below the melting point, hydrolysis of PET catalyzed by liquid acid in water proceeds at the solid–liquid interface. To increase hydrolysis efficiency, high concentrations of liquid acid are usually used to roughen the surface of PET solid, which produces a high effective reaction area. However, increasing the concentration of liquid acid has many drawbacks such as facility contamination, environmental pollution, and high cost of the subsequent separation of product and catalyst. Consequently, design of active, sustainable, and environmentally friendly catalysts and the corresponding PET hydrolysis reactions is a promising emerging field.
Recently, ionic liquids have drawn much attention as functional phase transfer catalysts and been used in the degradation of PET with excellent activity and reusability.17–19 However, despite achieving almost complete hydrolysis, the extremely high cost of the complex ingredients of ionic liquids has impeded their industrial use. Solid acids have advantages such as high acid strength, lower facility contamination, easy separation from the reaction medium, and reusability. Nevertheless, few applications of solid acids for PET hydrolysis have been reported,20 which may be because of the difficulty in the mass transport between solid PET and the solid acid at temperatures below the melting point of PET. Therefore, a green, efficient reaction medium is necessary to achieve sufficient contact among PET, water, and the active acid sites on the catalyst. Supercritical fluids have excellent dissolving and transfer capacity because of their unique properties such as liquid-like density, gas-like viscosity, and high diffusion. Carbon dioxide, which has desirable properties such as a low mild critical point (Tc = 31.4 °C, Pc = 7.38 MPa), non-toxicity, easy availability, chemical inertness, and an environment benign nature, is widely used in the modification of PET. Supercritical CO2 (SCCO2) shows remarkable sorption and diffusion into semi-crystalline polymers, and dissolving CO2 in PET alters the properties of PET in glass and rubbery states by swelling the PET matrix to expand the free volume for the mobility of chain segments,21,22 depressing the glass transition temperature and melting point,23,24 and inducing crystallization of amorphous PET.25
In our previous work,20 we synthesized sulfated titania (SO42−/TiO2) and used it as a solid superacid to achieve the complete catalytic hydrolysis of PET in supercritical CO2. The reaction was conducted in the temperature range of 100–160 °C with a partial pressure range of CO2 of 8–15 MPa for different durations to investigate the kinetics and hydrolysis mechanism. A supercritical phase containing water vapor and CO2 was generated in this reaction system, and water molecules adsorbed chemically on the surface of the SO42−/TiO2 catalysts, converting Lewis acid sites into Brønsted acid sites. The effect of SCCO2 in this system can be summarized as follows: (i) swelling the PET matrix to create sufficient free volume for the bulk reaction; and (ii) transporting water and hydronium ions (Brønsted acid sites) into the amorphous regions of PET matrix, resulting in the hydrolysis occurring preferentially in the amorphous regions of the surface and bulk of the PET matrix. However, a further study showed that SO42−/TiO2 solid acid catalysts release the sulfate species coordinated to the surface in the presence of water vapor, decreasing the catalyst acidity and activity. These results suggest that the hydrolysis of PET requires solid acid catalysts with higher hydrothermal stability.
Since the pioneering work of Hino and Arata,26 WOx/MxOy has received considerable attention as a strong solid acid. Despite its lower acidity compared with the SO42−/MxOy solid superacid, the increased thermal stability of WOx/MxOy in hydrogen, oxygen, and water means that it is an alternative catalyst in reaction systems requiring strong acid sites such as for isomerization of light alkanes,27 alcohol dehydration,28 and esterification and transesterification reactions.29 Many studies have attempted to correlate the structure of WOx/MxOy with its acidity and catalytic performance, and the results suggest that the active structural models depend on the catalyst and reaction system. Several structural models of the WOx species generated on the surface of WOx/MxOy catalysts have been proposed, including monotungstate (tetrahedral or octahedral coordination), polytungstate, nano-WOx clusters, and well-ordered crystalline WO3, depending on the surface W atom density.30,31 However, these structural models of the catalytically active sites remain controversial. Some researchers32–34 synthesized a series of WOx/MxOy (M = Ti, Zr, Al) by the incipient wetness impregnation method, and investigated the relationship between acidity, surface structure, and catalytic activity by in situ Raman and infrared spectroscopy. The results indicated a direct relationship between the abundance of polytungstate species, the number of strong Brønsted acid sites, and the catalytic activity. Zhou et al.27,35 reported the high-angle annular dark-field imaging of WOx/ZrO2 catalysts with an aberration-corrected analytical electron microscope, which allowed direct imaging of the various species present on the surface. After comparing the WOx species in samples showing high and low activities, the active catalytic site was identified as being associated with WOx clusters 0.8–1 nm in diameter and intermixed with a small amount of ZrOx, namely subnanometer Zr–WOx clusters. Through a combination of catalytic and Raman spectroscopic studies, Hammond et al.36 proved that instead of monotungstate or polytungstate species, crystalline WO3 is the most active and stable phase for olefin epoxidation with H2O2, and subsequent optimal activity was found for crystalline WO3 nanoparticles prepared by flame aerosol technology. Additionally, it was reported37–39 that incorporating doped or mixed metal ions M(1) into the support metal ions M(2) formed charge-imbalanced M(1)–O–M(2) bonds, generating acid sites. To summarize, the acidic power and catalytic activity of tungsten-promoted metal oxides may originate from (i) the inductive effect of the WOx species with a special active structure on the surface, (ii) crystalline WO3 on the surface behaving as a strong solid acid, or (iii) charge imbalance of the lattice structure caused by framework substitution of W6+ with host metal ions. The structural model for active catalytic sites and the catalytic mechanism are linked to the catalytic reaction system. However, WOx/MxOy has not been used in the hydrolysis of PET, and investigating the structural model for active catalytic sites of WOx/MxOy and the corresponding catalytic mechanism in PET hydrolysis will be important in guiding the design and development of green, efficient hydrolysis of waste polyesters.
In this work, a catalytic reaction system involving supercritical CO2 as reaction medium was developed. Tungsten-promoted titania solid acid catalysts were prepared by a hydrothermal method and used in the hydrolysis of PET. The results indicated complete hydrolysis was achieved and high-purity TPA, EG, and DEG obtained. The structure of surface WOx species was controlled by adjusting the tungsten loading. X-ray diffraction (XRD), laser Raman spectroscopy, and high resolution transmission electron microscopy (HR-TEM) were used to investigate the active structure of WOx species in this reaction system. NH3-temperature programmed desorption (NH3-TPD), ion-exchange titration, and H2-temperature programmed reduction (H2-TPR) were conducted to analyze the surface acidity and reducibility of the catalysts. Based on these results, we deduced the relationship between the active structure, surface acidity, and catalytic performance for PET hydrolysis. Additionally, scanning electron microscopy (SEM) was used to observe the morphologies of the surface and cross section of the PET flakes at different degrees of hydrolysis to investigate the PET hydrolysis mechanism.
2 Experimental
2.1 Materials
Chemically pure cetyltrimethyl ammonium bromide (CTAB, 99.0%), ammonium metatungstate hydrate (AMT, 99.0%), and titanium tetrabutoxide (TBOT, 96.0%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Analytically pure aqueous ammonia (NH3·H2O, 25.0–28.0%), ethanol (C2H5OH, 99.7%), and dimethyl sulfoxide (DMSO, 99.0%) were obtained from Shanghai Lingfeng Chemical Reagent Co., Ltd. Carbon dioxide (CO2, 99.9%) was supplied by Hukang Gas Co., Ltd. Deionized water was obtained by doubly distilling water pretreated by reverse osmosis.Waste transparent PET bottles (Mn ≈ 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1) were obtained from Coca-Cola Company. The bottles were shredded into sheets of 2 × 2 × 0.4 mm and cleaned sequentially with detergent, ethanol and deionized water under ultrasonication for 1 h, and then vacuum dried at 50 °C for 12 h.
100 g mol−1) were obtained from Coca-Cola Company. The bottles were shredded into sheets of 2 × 2 × 0.4 mm and cleaned sequentially with detergent, ethanol and deionized water under ultrasonication for 1 h, and then vacuum dried at 50 °C for 12 h.
2.2 Preparation of catalysts
WOx/TiO2 solid acid catalysts with various W/Ti molar ratios (designated as RW/Ti) were prepared by a hydrothermal method. In a typical procedure, cetyltrimethyl ammonium bromide (5.65 g) was dissolved in anhydrous ethanol (35.6 g). Titanium tetrabutoxide (17 g) was added and the mixture was stirred in a custom-made beaker with four vertical baffles at 30 °C for 1 h to obtain a homogeneous transparent solution. In another beaker, an appropriate amount of ammonium metatungstate hydrate was dissolved in deionized water (3.6 g), and then pumped into the titanium solution at 0.5 mL min−1 under constant and vigorous mechanical agitation. The opaque suspension was further stirred at 30 °C for 3 h, and then the pH of the suspension was adjusted to 11 with 0.028 g g−1 aqueous ammonia. The resulting slurry was transferred into a Teflon-lined autoclave and aged at 100 °C for 24 h. After drying at 70 °C for 12 h, the solids were put in a muffle furnace and heated to 500 °C at a rate of 2 °C min−1, and kept at 500 °C for 6 h to generate the WOx/TiO2 solid acid catalyst, which was named x W–Ti, where x is 0.00, 0.05, 0.10, 0.20, 0.30, 0.40, or 0.50, indicating the W/Ti molar ratio, depending on the tungsten loading percentage. In addition, sulfated titania was also prepared following the procedure reported in our previous work,20 and pure WO3 was obtained by heating ammonium metatungstate hydrate at 500 °C for 6 h under air.2.3 Characterization of catalysts
Powder X-ray diffraction (XRD) analyses were performed on a diffractometer (D/MAX2550VB/PC, Rigaku) using Cu Kα emission radiation and a carbon monochromator. The average crystallite size was calculated by using the Scherrer equation, and the crystalline phase was quantitatively analyzed by the Rietveld refinement method.Nitrogen physisorption was used to measure the specific surface areas, pore volumes and pore sizes of the catalysts with a static volumetric instrument (ASAP2010, Micromeritics, USA). The adsorption–desorption isotherms were measured at −197 °C after heat pretreatment under vacuum for 3 h at a temperature of 300 °C. The pore size distribution and pore volume were calculated with the BJH method based on the desorption isotherm, and surface area was calculated by using the BET method based on the adsorption isotherm.
Raman spectra were collected with a Raman spectrometer (InVia, Renishaw) equipped with a Nd-YAG laser operating at 514 nm and a CCD detector. The laser beam was focused through microscope objective lenses (100×) to a 1 μm spot on the sample. The intensities of the ν(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bands at 984 cm−1 were determined by using a linear background for the overall spectral region. After normalizing the spectra with respect to the anatase band at 636 cm−1, band areas were obtained by curve-fitting with a Gaussian profile, keeping the position and FWHM nearly constant.
O) bands at 984 cm−1 were determined by using a linear background for the overall spectral region. After normalizing the spectra with respect to the anatase band at 636 cm−1, band areas were obtained by curve-fitting with a Gaussian profile, keeping the position and FWHM nearly constant.
High-resolution transmission electron microscopy (HR-TEM) images of the catalyst samples were recorded on a transmission electron microscope (JEM-2100 LaB6, JEOL) equipped with an EDS detector operating at 200 kV.
NH3-temperature programmed desorption (NH3-TPD) measurements were conducted on an Autochem II 2920 instrument (Micromeritics, USA) with a thermal conductivity detector (TCD) used for continuous monitoring of desorbed ammonia. The sample (0.1 g) was pretreated at 500 °C for 1 h in a flow of ultra-pure He gas (50 mL min−1) and saturated with pure anhydrous ammonia gas at 50 °C for 0.5 h. After sweeping with He (50 mL min−1) at 50 °C for 1 h to remove the physisorbed ammonia, the sample was heated from 50 °C to 800 °C at a rate of 10 °C min−1. H2-temperature programmed reduction (H2-TPR) was performed on the same reaction system. Prior to the analysis, a fresh sample was treated at 500 °C for 1 h in a stream of O2/He (1% v/v, 50 mL min−1). Then the sample was heated from 50 to 1000 °C at a 10 °C min−1 ramp in a stream of H2/N2 (10% v/v, 50 mL min−1), and the hydrogen consumption was detected by the TCD. The mass of the catalyst samples varied from 0.013 to 0.100 g so that approximately the same molar amount of WO3 was present.
The number of Brønsted acid sites was determined via a titration experiment involving an ion-exchange step,40 in which the catalyst sample (0.1 g) was added to 0.5 mol L−1 aqueous NaCl solution (15 mL). After 24 h of ion exchange under stirring at 30 °C between the catalyst H+ and Na+ in solution, the liquid was filtered off and titrated with a 0.1 mol L−1 aqueous NaOH solution.
2.4 Catalytic hydrolysis of waste bottle PET
:0.1:10, and the initial mass of the PET (m0,PET) and catalyst (mCat) were measured. A custom-made buffer tank with a heater was connected to the vessel to supply high pressure CO2. The pressure in the vessel was measured by a pressure transducer, and the temperature of the fluid in the vessel was monitored by a thermocouple and controlled by a matched automatic heater with a temperature accuracy of ±0.1 °C. At the end of each run, the heater was unloaded and the vessel was allowed to cool to room temperature. After releasing the CO2, the two cells were removed for product separation and analysis.
 | ||
| Fig. 1 Schematic diagram of the reactor system for PET hydrolysis in SCCO2. (A) CO2 cylinder; (B) buffer tank; (C) vacuum pump; (D1 and D2) thermocouples; (E) pressure monitor; (F) heater (oil bath); (G) pressure cell; (H) holder; (I) pressure transducer; (J) agitator; (K1–K6) needle valves; (O1 and O2) safety valves. | ||
 | (1) |
The filtered solid was washed three times with DMSO to dissolve the TPA monomer, the residual PET and catalysts were filtered and dried under vacuum at 120 °C for 6 h to obtain the total mass of the mixture of residual PET and catalyst (mt,PET/Cat), and then the mass of residual PET and the degree of PET hydrolysis were calculated by eqn (2) and (3).
| mt,PET = mt,PET/Cat − mCat | (2) |
 | (3) |
TPA was precipitated by adding deionized water to the TPA/DMSO solution. After filtering and drying under vacuum at 120 °C to a constant weight, the recovered TPA was weighed, and the mass yield of TPA was calculated by eqn (4).
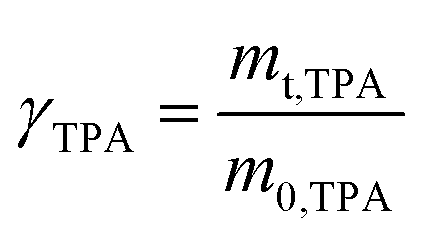 | (4) |
After the reaction and separation steps, the catalysts were reactivated at 120 °C under air for 6 h (with some residual PET) and were reused for another PET hydrolysis reaction to evaluate the reusability of the catalysts.
3 Results
3.1 Physicochemical properties of the tungsten-promoted titania
The crystal structure properties of the tungsten-promoted titania samples with various tungsten loadings were characterized by XRD (Fig. 2). All samples had the anatase crystal structure regardless of tungsten loading; however, the diffraction peaks characterizing anatase broadened and decreased in intensity with increased tungsten loading. This is consistent with several reports indicating that the addition of the tungsten decreases crystallinity and increases crystal transform temperature because WOx species hinder crystal growth.30,41,42 The Scherrer crystallite size of the anatase (Table 1) decreased with increased tungsten loading, and the calculated crystallite size decreased continuously from 14.6 (0.00 W–Ti) to 1.3 nm (0.50 W–Ti), owing to either the framework substitution of Ti4+ with W6+ or the accumulation of WOx species on the grain boundaries. There was no distinct tungsten oxide diffraction peak observed in samples with RW/Ti of up to 0.30 mol mol−1. However, at high tungsten loadings (0.40 W–Ti and 0.50 W–Ti), a broad diffraction peak emerged as a shoulder on the (101) anatase diffraction peak at 25.3°, which is characteristic of crystalline WO3; WO3 has its three most intense diffraction peaks in this region ((002), (020), (200)). Considering that the tungsten loadings of all samples were above the XRD detection limit, the absence of WO3 diffraction peaks was attributed to an amorphous surface WOx structure or the incorporation of W6+ into the anatase lattice replacing Ti4+ to form a W–O–Ti bond because the ionic radii of W6+ (0.600 Å) and Ti4+ (0.605 Å) are similar.43,44 | ||
| Fig. 2 XRD patterns of tungsten-promoted titania with various tungsten loadings. (A) 0.00 W–Ti; (B) 0.05 W–Ti; (C) 0.10 W–Ti; (D) 0.20 W–Ti; (E) 0.30 W–Ti; (F) 0.40 W–Ti; (G) 0.50 W–Ti. | ||
| Catalyst | dA, nm | a, Å | c, Å | Cell volume, Å3 |
|---|---|---|---|---|
| TiO2 | 14.6 | 3.7850 | 9.5224 | 136.27 |
| 0.05 W–Ti | 8.0 | 3.7876 | 9.5157 | 136.51 |
| 0.10 W–Ti | 6.3 | 3.7854 | 9.5138 | 136.32 |
| 0.20 W–Ti | 5.2 | 3.7850 | 9.5217 | 136.41 |
| 0.30 W–Ti | 2.3 | 3.7865 | 9.5199 | 136.49 |
| 0.40 W–Ti | 1.5 | 3.7862 | 9.5239 | 136.53 |
| 0.50 W–Ti | 1.3 | 3.7857 | 9.5244 | 136.50 |
The lattice distortion of the mixed metal oxide caused by lattice substitution is reflected in the lattice parameters, which can be calculated from the XRD data by the Rietveld method.45,46 Kim et al.47 synthesized anatase nanocrystals containing lattice and surface doped tungsten by the hydrothermal method. The a-parameter, c-parameter and cell volume all increased monotonically as the content of W doped into the titania lattice increased (a-parameter increased from 3.7874 (RW/Ti = 0.014 mol mol−1) to 3.8009 Å (RW/Ti = 0.126 mol mol−1); c-parameter increased from 9.4807 (RW/Ti = 0.014 mol mol−1) to 9.4986 Å (RW/Ti = 0.126 mol mol−1)). After saturation was reached, additional W generated surface WOx species exerted pressure in the a-direction, resulting in the shrinkage of the c-axis to maintain the unit cell volume, because the concentration of the lattice doping tungsten is constant in the saturation range (a-parameter increased to 3.8153 Å (RW/Ti = 0.353 mol mol−1); c-parameter decreased to 9.4459 Å (RW/Ti = 0.353 mol mol−1)). Fuerte48 and Fernández-García49 synthesized W–Ti mixed oxide nanoparticles by the reverse micelle technique, and the monotonic dependence of lattice parameters on the W content in the titania lattice was observed. Riboni et al.46 prepared a series of WO3–TiO2 photocatalysts by the impregnation method and analyzed the crystal structure. W existed as surface WOx species and the lattice parameters of titania remained almost constant despite the variation of W loading. In the present study, according to the lattice parameters in Table 1, a non-monotonic dependence on tungsten loading was observed, indicating limited insertion of W into the anatase lattice and that amorphous WOx species or crystalline WO3 mainly formed on the catalyst surface.
The textural properties of the tungsten-promoted titanias with RW/Ti of 0.00, 0.10, 0.30, and 0.50 mol mol−1 were characterized by N2 physisorption (Fig. 3 and Table 2). According to IUPAC nomenclature, all samples exhibited a typical type IV adsorption isotherm, indicating a mesoporous structure, followed by a H1 type hysteresis loop, suggesting the presence of cylindrical mesopores. The BJH desorption pore diameters of tungsten-promoted titanias of 5.5–6.0 nm were smaller than that of pure titania of 8.8 nm. The BET surface area of the pure titania was 44 m2 g−1, which increased dramatically with increased tungsten loading, from 126 m2 g−1 for 0.10 W–Ti samples to 172 m2 g−1 for 0.30 W–Ti samples. However, the BET surface area decreased to 87 m2 g−1 as RW/Ti increased to 0.50 mol mol−1. A similar trend was also observed for the pore volume of the samples, which increased gradually from 0.12 (0.00 W–Ti) to 0.29 cm3 g−1 (0.30 W–Ti), and then decreased slightly to 0.17 cm3 g−1 (0.50 W–Ti). Compared with pure titania, the higher BET surface area and pore volume of tungsten-promoted catalysts with increased tungsten loading was attributed to formation of amorphous WOx species on the surface and grain boundaries preventing crystal growth and grain sintering during calcination. The three-dimensional crystalline WO3 formed at high tungsten loadings (0.50 W–Ti) covering the inner surface may have occupied the pore structure, decreasing the surface area and pore volume.
 | ||
| Fig. 3 (A) N2 sorption isotherms and (B) pore size distributions of tungsten-promoted titania with various tungsten loadings. | ||
| Catalyst | SBET, m2 g−1 | P, nm | V, cm3 g−1 |
|---|---|---|---|
| 0.00 W–Ti | 44 | 8.8 | 0.12 |
| 0.10 W–Ti | 126 | 5.5 | 0.20 |
| 0.30 W–Ti | 172 | 6.0 | 0.29 |
| 0.50 W–Ti | 87 | 5.7 | 0.17 |
The nature of WOx species growing on the titania support was investigated by Raman spectroscopy (Fig. 4). There are several possible WOx structures, such as tetrahedral or octahedral coordination, polytungstate species, nonstoichiometric tungsten oxides, and multiple oxidation states,30,33,41 depending on the synthesis method, tungsten loading, and calcination temperature and duration. All tungsten-promoted titania samples with various tungsten loadings exhibited titania-related vibrations at 396, 516 and 638 cm−1, characteristic of the anatase structure. The anatase peaks broadened and decreased in intensity with increased tungsten loading, resulting from the decrease in crystallite size and crystallinity because surface WOx species prevent crystal growth. No distinct peaks at 276, 719 and 808 cm−1 corresponding to crystalline WO3 were observed at low tungsten loadings. However, two peaks emerged at 719 and 808 cm−1 adjacent to the tail of the broad anatase peak at 638 cm−1 for the tungsten-promoted samples with RW/Ti of 0.40 and 0.50 mol mol−1. The peaks were attributed to the W–O stretching mode of crystalline WO3. Additionally, a peak at 984 cm−1 was observed for all tungsten-promoted titania samples. There were no peaks related to crystalline WO3 around 984 cm−1. However, WOx species had characteristic peaks in the 800–1000 cm−1 region attributed to the symmetric WO stretching mode of surface dispersed two-dimensional WOx species, and the peak position depended on the dispersion state and the condensation degree of the WOx species.
 | ||
| Fig. 4 Raman spectra of tungsten-promoted titania. (A) 0.00 W–Ti; (B) 0.05 W–Ti; (C) 0.10 W–Ti; (D) 0.20 W–Ti; (E) 0.30 W–Ti; (F) 0.40 W–Ti; (G) 0.50 W–Ti. | ||
It has been reported30 that within its monolayer dispersion capacity, WOx species are highly dispersed as isolated tetrahedral species (WO4) at low tungsten coverage, and as tungsten loading increases, an octahedrally coordinated WOx species (WO6) appears. The ratio of WO6 to WO4 increases gradually with increased tungsten coverage, and the WO4 and WO6 units meet and generate a cross-linked polytungstate structure, the degree of condensation of which increases with further increasing tungsten coverage. Once the monolayer coverage is reached, crystalline WO3 is formed. The corresponding peak in the Raman spectra characteristic of the WO shifts from 958 to 984 cm−1 with increased tungsten loading. The peak at 958 cm−1 is assigned to the symmetric stretch of the WO bond within the distorted WO4 species, and the peak at 984 cm−1 belongs to polytungstate, which is the most condensed dispersion state of all the WOx structures. However, in the present work, all the tungsten-promoted titania samples showed the WO peak at 984 cm−1 with no distinct shift, indicating the presence of polytungstate on the surface, because all the tungsten-promoted titania samples had a high tungsten coverage.
The WOx species on the surface of all the samples existed as polytungstates and the Raman results suggested the WOx species were similar; therefore, the differences among these polytungstates with various tungsten coverages were investigated. The WOx species formed on the titania supports were observed by HR-TEM (Fig. 5). The uniform lattice fringes of the anatase crystalline structure were observed in all samples without major variations in the fringe spacing of the tungsten-promoted titania, indicating little substitution of Ti4+ with W6+. The fringe spacing of 0.238 and 0.352 nm were indexed to the (004) and (101) plane of anatase, respectively. The 0.00 W–Ti samples consisted of micrometer-sized particles with smooth surface morphology, whereas nanoparticles aggregates with rough surfaces were observed in tungsten-promoted titania samples. Compared with 0.00 W–Ti samples, 0.10 W–Ti samples had black spots with diameters of less than 1 nm on the surface (indicated by yellow arrows and yellow circles in Fig. 5). These spots were identified as surface polytungstate species with a two-dimensional network structure with several tungsten atoms linked by oxygen bridging bonds.41 For 0.30 W–Ti samples, the size of these spots increased remarkably and many WOx clusters with diameters of 3–5 nm were observed, indicating a much higher degree of condensation of the polytungstate species. However, as the tungsten coverage increased further, the large WOx clusters disappeared and crystalline WO3 emerged (Fig. 5(D)). In the 0.50 W–Ti sample, the fringe spacing of 0.385 nm was attributed to the (002) plane of crystalline WO3, and black spots assigned as polytungstate with diameters of 1–2 nm were also observed. In summary, the Raman spectra and HR-TEM results showed that polytungstates were generated at low tungsten coverage with a low degree of condensation, which increased with increasing tungsten coverage. At moderate tungsten coverage, large WOx clusters with the highest degree of condensation were formed, and they transformed into crystalline WO3 as the tungsten coverage increased further.
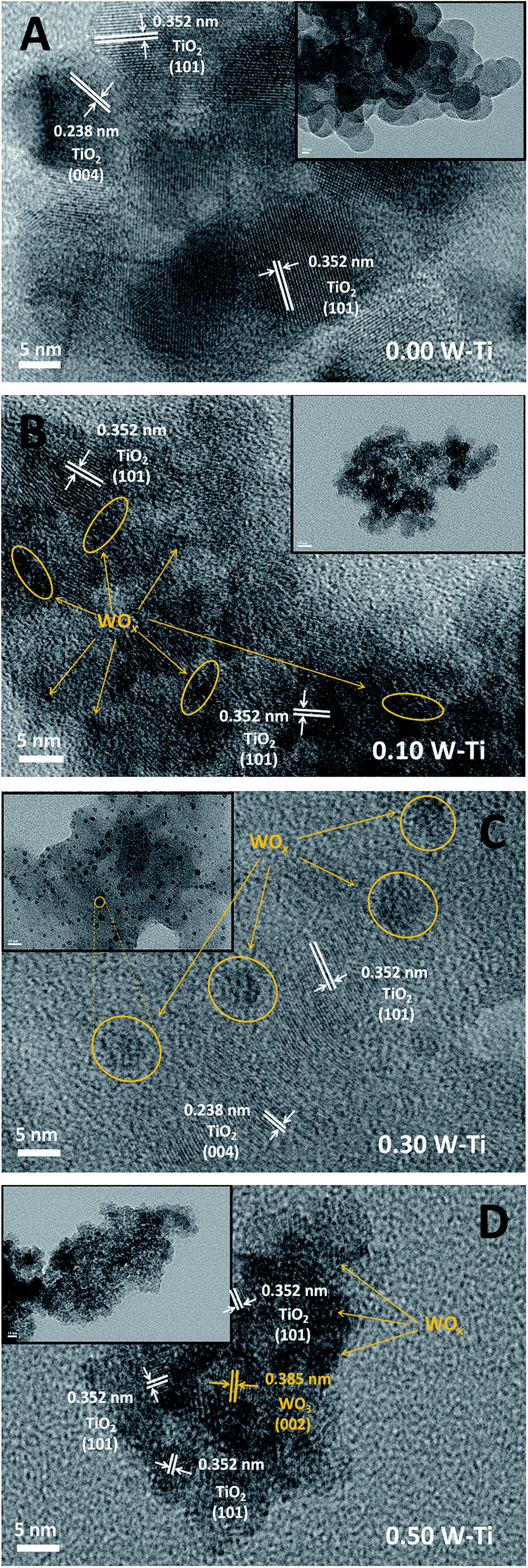 | ||
| Fig. 5 HR-TEM images of (A) 0.00 W–Ti, (B) 0.10 W–Ti, (C) 0.30 W–Ti and (D) 0.50 W–Ti. Yellow arrows indicate some surface nano-WOx clusters, and yellow circles highlight the regions with high numbers of nano-WOx clusters in close proximity and some large WOx clusters. | ||
The active WOx species formed on the titania support endow the tungsten-promoted titania samples with high acidity. Active WOx species have high electronegativity and pull the electron cloud from the titania support, producing electron-deficient Ti4+ centers that function as Lewis acid sites, or hydroxyl groups attached to the Ti4+ or W6+ centers that function as Brønsted acid sites. The surface acidic properties, such as total acid content and the distribution of acid strength of the tungsten-promoted titania samples, were investigated by NH3-TPD characterization (Fig. 6). For the tungsten-promoted titania samples with RW/Ti of 0.30 mol mol−1 or less, the total acidity increased with increasing tungsten loading. Four desorption peaks corresponding to acid sites of different acid strength were observed: a desorption peak assigned to weak acid sites at 140–160 °C; a wide peak assigned to medium acid sites at 260–320 °C; and two peaks belonging to strong acid sites at 400–500 °C and above 600 °C. However, as tungsten loading increased further, the total acidity decreased slightly and the strong acid sites above 600 °C disappeared, resulting from the decrease in surface area and conversion of active WOx species into inactive crystalline WO3 at high tungsten loading.
 | ||
| Fig. 6 NH3-TPD curves of the tungsten-promoted titania with different W loadings. | ||
The hydrolysis of PET is a typical Brønsted acid-catalyzed reaction. The active WOx species on the titania support create Brønsted acid sites, and the proportion of Brønsted acid sites to Lewis acid sites increases with increasing tungsten loading, which has been confirmed by the in situ pyridine-FTIR technique.33,41,50 In the present work, the number of total Brønsted acid sites was determined by titration experiments. Fig. 13 shows that the number of Brønsted acid sites increased continuously from 0.296 mmol g−1 for 0.05 W–Ti to 1.43 mmol g−1 for 0.30 W–Ti, followed by a slight decrease to 1.31 mmol g−1 for 0.50 W–Ti.
EG, which was produced during PET hydrolysis, could chemically reduce WOx species. To investigate the reduction of the catalysts, H2-TPR experiments were performed (Fig. 7). Within the temperature range covered by the experiments (room temperature to 1000 °C), no reduction peak was observed for 0.00 W–Ti, whereas pure WO3 exhibited three reduction peaks at 768, 861 and above 1000 °C. Similarly, all the tungsten-promoted titania samples exhibited three reduction peaks corresponding to different steps of the reduction of W6+ on the surface, at a lower reduction temperature than crystalline WO3. The first small reduction peak centered around 500–600 °C was attributed to the first reduction step of W6+ (Step I: WO3 → WO2.9), whereas the other broad reduction peak at 600–800 °C was related to the second reduction step (Step II: WO2.9 → WO2). The incomplete peak above 1000 °C was attributed to the complete reduction of W6+ (Step III: WO2 → W).31,51 However, with increasing tungsten loading, the reduction peaks shifted gradually to a lower reduction temperature. The temperature of reduction Step I dropped from 515 (0.05 W–Ti) to 504 °C (0.30 W–Ti), and the temperature of Step II shifted from 800 (0.05 W–Ti) to 629 °C (0.30 W–Ti). These results indicate that the reducibility of the polytungstate species increased with its condensation degree; namely, the larger and more interconnected WOx clusters formed at high tungsten loading were easier to reduce than the smaller clusters formed at low tungsten loading. The reduction peaks shifted back to a higher temperature as tungsten loading increased further, for 0.50 W–Ti. The reduction temperature of Steps I and II increased to 565 and 652 °C, respectively, because of the formation of crystalline WO3, which has a lower reducibility.
 | ||
| Fig. 7 H2-TPR profiles of tungsten-promoted titania. (A) 0.00 W–Ti; (B) 0.05 W–Ti; (C) 0.10 W–Ti; (D) 0.20 W–Ti; (E) 0.30 W–Ti; (F) 0.40 W–Ti; (G) 0.50 W–Ti; (H) WO3. | ||
3.2 Catalytic hydrolysis of PET in supercritical CO2
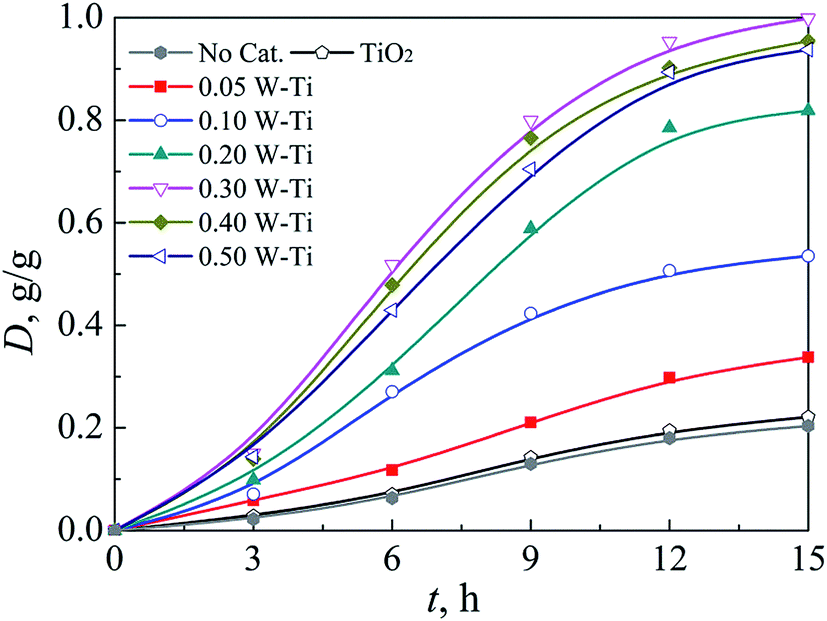 | ||
| Fig. 8 Degree of PET hydrolysis at 160 °C and 15 MPa over tungsten-promoted titania solid acid catalysts with various W loadings for different reaction durations. | ||
The corresponding mass yield of the products TPA, EG, and DEG are shown in Fig. 9. At 0–6 h, the mass yields of TPA and EG were slightly lower than the corresponding hydrolysis degree, probably due to mass loss during separation and analysis. The mass yield of EG was lower than that of TPA after 6 h, and the gap increased as the hydrolysis reaction continued. DEG was detected and its mass yield increased with reaction duration. DEG is derived from intra-molecular dehydration of EG catalyzed by the TPA generated or by concentrated liquid acid, such as sulfuric acid in some traditional PET hydrolysis systems.15,52 However, in our SCCO2–PET reaction system, the tungsten-promoted titania catalyzed the dehydration of EG to DEG instead of TPA.20.
 | ||
| Fig. 9 Mass yield of TPA, EG and DEG produced by PET hydrolysis at 160 °C and 15 MPa over tungsten-promoted titania solid acid catalysts with various W loadings for different reaction durations. | ||
The mass yield of DEG obtained by hydrolysis at 160 °C and 15 MPa for 15 h depended on the tungsten loading of the catalysts used. The yield increased from 0 for 0.05 W–Ti to 0.064 g g−1 for 0.30 W–Ti, whereas it decreased to 0.033 g g−1 for 0.50 W–Ti, which was probably related to the reducibility of the catalysts. Iglesia and colleagues53 investigated the structure–activity relationship of WOx/ZrO2 in the dehydration of alcohols, and confirmed that the alcohol reduced the polytungstate domains and formed Hδ+ acid centers via dissociation of O–H bonds. The Hδ+(WO3)nδ− species generated were stabilized by the polytungstate species and behaved as Brønsted acid sites. These in situ generated temporary acid sties catalyzed dehydration much more effectively than permanent acid sites, namely the acid OH groups on the support or the WOx species. In the present work, the TPR and HR-TEM results indicated that increasing the tungsten loading increased the reducibility of the WOx species, which promoted the generation of Hδ+ and increased the size of WOx clusters. These changes increased the dispersion of the extra electron density transferred to the WOx species and stabilized the acidic sites.54 Consequently, the mass yield of DEG increased with increasing tungsten loading in the RW/Ti range of 0.05–0.30 mol mol−1. Thomas et al.33 also attributed the higher activity of WOx/TiO2 compared with WOx/ZrO2 to the greater reducibility of the WOx species. However, the formation of WO3 decreased the mass yield of DEG because the WO3 crystallites blocked access to WOx and resulted in the formation of oxygen-deficient WO3−x species that do not delocalize the electron density required to stabilize Hδ+ formed from alcohol reactants.55
The tungsten-promoted titania materials were white or light yellow, and became darker with increased tungsten loading. However, the catalysts turned blue after each hydrolysis reaction, and after treatment at 120 °C under air for 6 h, they changed back to white or light yellow. Despite the desirable reusability, the change in the chemical state of the surface WOx species during catalytic hydrolysis revealed by the color change should be studied. H2-TPR and NH3-TPD were conducted to study the acidity and reducibility of the fresh, used and reactivated 0.30 W–Ti samples further, and the results are shown in Fig. S2 and S3.† The TPR results showed that the peak area of the used 0.30 W–Ti catalyst was smaller than that of the fresh catalyst, indicating that WOx on the surface of the used catalysts was partially reduced. However, after washing and reactivation, the shape and area of the reduction peak become similar to the fresh catalyst, suggesting that the partially reduced WOx was fully re-oxidized during reactivation. Additionally, the NH3-TPD profiles revealed that the acidity of 0.30 W–Ti decreased from 0.0374 to 0.0322 mmol NH3 per g after being used once, although it increased back to 0.0368 mmol NH3 per g after reactivation, whereas the position and shape of the desorption peaks remained nearly constant. The weaker acidity of the used catalysts was attributed to the decrease in electronegativity of the partly reduced polytungstate species, which led to weaker pulling of the electrons from the titania support.
 | ||
| Fig. 10 Evaluation of the reusability of 0.30 W–Ti compared with sulfated titania at 160 °C and 15 MPa for 12 h. | ||
 | ||
| Fig. 11 Morphology of the surface of the PET flakes with various reaction durations and hydrolysis degrees. (A) Original; (B) t = 3 h, D = 0.1503 g g−1; (C) t = 6 h, D = 0.5182 g g−1; (D) t = 9 h, D = 0.7991 g g−1; (E) t = 15 h, D = 0.9921 g g−1. | ||
 | ||
| Fig. 12 Morphology of the surface of the PET flakes with various reaction durations and hydrolysis degrees. (A) Original; (B) t = 3 h, D = 0.1503 g g−1; (C) t = 6 h, D = 0.5182 g g−1; (D) t = 9 h, D = 0.7991 g g−1; (E) t = 15 h, D = 0.9921 g g−1. | ||
The original PET, swelled PET and residual PET after catalytic hydrolysis were characterized by XRD to investigate the variation of crystallinity of PET during hydrolysis (Fig. S4†). All samples were washed with DMSO to remove EG, DEG and TPA. The original PET exhibited one wide diffraction peak at 25.5°, which increased in intensity after swelling with SCCO2 at 160 °C and 15 MPa for 9 h, and a new peak emerged at 22.6° for the swelled PET, suggesting that SCCO2 induced crystallization and increased the crystallinity of PET. However, after PET was hydrolyzed under the same conditions and catalyzed with 0.30 W–Ti, the residual PET exhibited diffraction peaks at 16.1°, 17.5°, 22.6° and 26.3°, which were characteristic of crystalline PET. Compared with swelled PET, the residual PET after catalytic hydrolysis had far higher crystallinity, indicating that hydrolysis occurred preferentially in the amorphous region of the PET and the crystalline region remained after hydrolysis for 9 h. The product had typical diffraction peaks for TPA crystallites at 17.3°, 25.1° and 27.9°, which are different from those of PET crystallites. The observation that the crystallinity of PET increased during hydrolysis was attributed to (i) the crystallization induced by SCCO2 and (ii) hydrolysis occurring preferentially in amorphous regions of PET.
4 Discussion
Characterization of the tungsten-promoted titania catalysts by XRD, Raman spectroscopy and HR-TEM indicated that the W phase was present as two-dimensional surface polytungstate species or three-dimensional crystalline WO3. Fig. 13 shows the evolution of the area of the ν(WO) band at 984 cm−1 associated with the polytungstate species in the Raman spectra, the number of Brønsted acid sites determined by ion-exchange titration experiments, and the average hydrolysis rate of PET over 15 h as functions of the W/Ti molar ratio (RW/Ti). A large increase in the amount of the polytungstate species was observed with increased W loading in the RW/Ti range of 0.05–0.30 mol mol−1, followed by a slight decrease in the RW/Ti range of 0.30–0.50 mol mol−1. Similar trends were also observed in the number of Brønsted acid sites and hydrolysis rate of PET, indicating a good correlation among the number of polytungstate species, the number of Brønsted acid sites and catalytic activity.
 | ||
| Fig. 13 Evolution of the area of the ν(WO) band at 984 cm−1 in Raman spectra, the number of Brønsted acid sites determined by ion-exchange titration experiment, and the average hydrolysis rate of PET within 15 h, as functions of W/Ti molar ratio. | ||
Fig. 14 shows the evolution of the intrinsic acidity and activity; namely, the number of Brønsted acid sites per W atom, the hydrolysis rate of PET per Brønsted acid site, and the hydrolysis rate of PET per W atom. A gradual increase in the number of Brønsted acid sites per W atom was observed from 0.578 (0.05 W–Ti) to 0.712 mol mol−1 (0.30 W–Ti), followed by a sharp decrease to 0.513 mol mol−1 (0.50 W–Ti). According to the HR-TEM results, the intrinsic acidic power showed a monotonic increase with the condensation degree of the polytungstate species, because the size of the WOx nanoclusters and the condensation degree of the polytungstate species increased with increasing tungsten loading in the RW/Ti range of 0.05–0.30 mol mol−1. However, in the RW/Ti range of 0.30–0.50 mol mol−1, as the inactive crystalline WO3 formed, the number of Brønsted acid sites per W atom decreased dramatically.
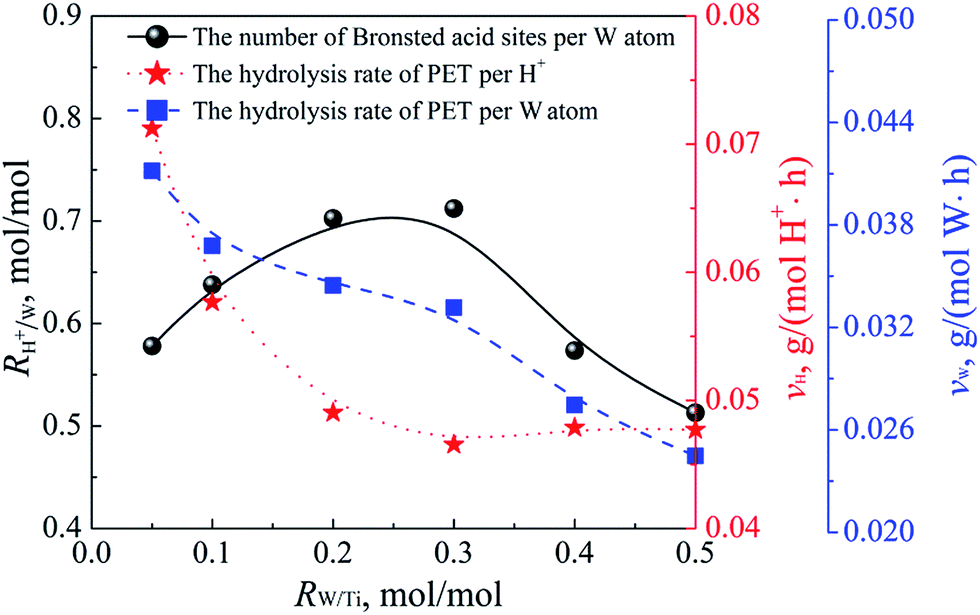 | ||
| Fig. 14 Number of Brønsted acid sites determined by ion-exchange titration experiment per W atom, the average hydrolysis rate of PET at 160 °C and 15 MPa for 15 h per W atom and per Brønsted acid site, as functions of W/Ti molar ratio. | ||
In contrast, the hydrolysis rate of PET per H+ decreased gradually until the RW/Ti reached 0.30 mol mol−1, and levelled off as the tungsten loading increased further. The number of the Brønsted acid sites determined by the titration method was not the same as the number of effective Brønsted acid sites with sufficient acidity to catalyze the reaction. Because the Brønsted acid sites generated by the increased tungsten loading may not be sufficiently acidic to hydrolyze PET, the hydrolysis rate of PET per H+ decreased despite the increase in the total number of Brønsted acid sites. Additionally, the strong mutual interaction between Brønsted acid sites and supercritical CO2 may also determine whether the acid site is effective or not, because the dissociated hydronium ions interact strongly with supercritical CO2 and are carried by supercritical CO2 to the surface or the bulk of the PET matrix. Similarly, the hydrolysis rate of PET per W atom showed a continuous monotonic decrease with the increase in RW/Ti owing to the generation of inactive Brønsted acid sites or the formation of inactive crystalline WO3.
The catalytic hydrolysis of PET over tungsten-promoted titania catalysts in supercritical CO2 can be divided into three steps. In the first step (0–3 h), supercritical CO2 swells the PET matrix to create sufficient bulk space, and hydrolysis occurs primarily on the surface, leading to a slow increase in hydrolysis degree. In the second step (3–9 h), the PET matrix is completely swollen, water molecules and effective Brønsted acid sites are carried by supercritical CO2 and penetrate the PET matrix, and the reaction occurs preferentially in the amorphous region of both the surface and bulk of PET matrix, leading to a sharp increase in hydrolysis degree with reaction time. In the third step (over 9 h), deactivation of the catalysts by partial reduction of the WOx species and crystallization induced by CO2 causes the increase in the hydrolysis degree to slow and level off gradually, especially for tungsten-promoted titania with higher activity.
As the surface WOx species were reduced, the acidity and activity decreased. Moreover, owing to the plasticization of CO2, the glass-transition temperature and crystallization temperature of PET decreased, which increased the mobility of the PET segments. The strong interaction between the CO2 and PET segments led to the formation of a regular chain structure, inducing crystallization. Because the regular chain structure is far more stable than a soft, random chain structure, the hydrolysis occurred in the amorphous regions of PET prior the crystalline region. At the end of hydrolysis, the anti-degradation properties of the residual crystalline region of PET also contributed to the slow hydrolysis rate in this step. At each reaction step, short-chain PET oligomers that are soluble in SCCO2 were generated. The oligomers were carried to the catalyst to be hydrolyzed further to TPA at the Brønsted acid sites, and thus some Brønsted acid sites that could not be carried by SCCO2 may still have played a role in PET hydrolysis. Similar mechanisms have been proposed for cellulose hydrolysis and cellulose conversion to small molecules like EG,56,57 where robust cellulose inert in most solvents was degraded by bi-functional catalysis involving unconventional media such as supercritical water or ionic liquids.58 Cellulose was first degraded by an in situ protic acid in the media, then the smaller, soluble molecules generated, such as oligosaccharides, disaccharides and monosaccharides, were catalytically degraded further to EG over the solid acid.
5 Conclusion
Tungsten-promoted titania solid acid catalysts were prepared by a hydrothermal method and used in the hydrolysis of waste bottle PET in supercritical CO2. PET was hydrolyzed completely at 160 °C and 15 MPa within 15 h. TPA, EG and DEG were obtained and no other by-products were detected. The catalysts were recycled and reused five times without noticeable performance loss. The active structural model of the catalysts and the PET hydrolysis mechanism were investigated, we drew the following conclusions.The tungsten phase existed as surface WOx species, and a direct correlation was observed among the amount of polytungstate species on the surface, the number of Brønsted acid sites and the catalytic activity during PET hydrolysis. Polytungstate species with a higher condensation degree endowed the catalysts with higher intrinsic acidity (number of Brønsted acid sites per W atom), and the polytungstate WOx species were easier to reduce as the degree of condensation increased.
The variation in the chemical state of WOx species via reduction with EG during hydrolysis decreased the permanent acidity of the catalysts, which determined the hydrolysis degree of PET, although it was refreshed by reactivation. The partial reduction of polytungstate also played a part in the formation of temporary acid sites, which determined the yield of DEG.
The PET hydrolysis mechanism over tungsten-promoted titania solid acid catalysts in supercritical CO2 can be described by three steps. Initially, the supercritical CO2 swells the PET and the reaction occurs primarily on the surface. Subsequently, supercritical CO2 carries hydronium ions (Brønsted acid sites) and water molecules into the amorphous regions of the swollen PET, and hydrolysis occurs both on the surface and in the bulk. Simultaneously, supercritical CO2 induces crystallization in the amorphous region. Finally, the stiff, tough crystalline regions begin to degenerate with a relatively low reaction rate until complete hydrolysis is achieved.
Based on our results, the catalytic system involving SCCO2 reported here is also suitable for the degradation of other macromolecules, such as cellulose, or for other chemically recycling methods for polyesters, including methanolysis and glycolysis, because SCCO2 interacts strongly with these polymers and shows excellent transporting capacity for the reactants.
Acknowledgements
The authors thanks for the support from Shanghai College Students Innovative Entrepreneurial Training Plan Program (S15001) and the assistance of Ye-Xin Du, Zhong-Wang Fu, Ao Xiao and Ren-Jie Tang.References
- ICIS Global PET meeting, http://www.icis.com/, accessed March 2014.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2008, 18, 8–25 CrossRef.
- H. Kurokawa, M.-A. Ohshima, K. Sugiyama and H. Miura, Polym. Degrad. Stab., 2003, 79, 529–533 CrossRef CAS.
- F. Chen, G. Wang, W. Li and F. Yang, Ind. Eng. Chem. Res., 2013, 52, 565–571 CrossRef CAS.
- A. Mittal, R. K. Soni, K. Dutt and S. Singh, J. Hazard. Mater., 2010, 178, 390–396 CrossRef CAS PubMed.
- D. Paszun and T. Spychaj, Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- R. López-Fonseca, M. P. González-Marcos, J. R. González-Velasco and J. I. Gutiérrez-Ortiz, J. Chem. Technol. Biotechnol., 2009, 84, 92–99 CrossRef.
- J. H. Jung, M. Ree and H. Kim, Catal. Today, 2006, 115, 283–287 CrossRef CAS.
- T. Yoshioka, M. Ota and A. Okuwaki, Ind. Eng. Chem. Res., 2003, 42, 675–679 CrossRef CAS.
- D. Z. Lixin Liu, L. An, H. Zhang and Y. Tian, J. Appl. Polym. Sci., 2005, 95, 4 Search PubMed.
- A. Ruvolo-Filho and P. S. Curti, Ind. Eng. Chem. Res., 2006, 45, 7985–7996 CrossRef CAS.
- D. A. S. Ravens, Polymer, 1960, 1, 375–383 CrossRef CAS.
- T. Yoshioka, T. Sato and A. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353–1355 CrossRef CAS.
- T. Yoshioka, T. Motoki and A. Okuwaki, Ind. Eng. Chem. Res., 2001, 40, 75–79 CrossRef CAS.
- T. Yoshioka, N. Okayama and A. Okuwaki, Ind. Eng. Chem. Res., 1998, 37, 336–340 CrossRef CAS.
- S. Mishra, A. S. Goje and V. S. Zope, Polym.-Plast. Technol. Eng., 2003, 42, 581–603 CrossRef CAS.
- F. Liu, X. Cui, S. Yu, Z. Li and X. Ge, J. Appl. Polym. Sci., 2009, 114, 3561–3565 CrossRef CAS.
- L. Zhang, J. Gao, J. Zou and F. Yi, J. Appl. Polym. Sci., 2013, 130, 2790–2795 CrossRef CAS.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568 RSC.
- X. K. Li, H. Lu, W. Z. Guo, G. P. Cao, H. L. Liu and Y. H. Shi, AIChE J., 2015, 61, 200–214 CrossRef CAS.
- J. von Schnitzler and R. Eggers, J. Supercrit. Fluids, 1999, 16, 81–92 CrossRef CAS.
- M. Champeau, J. M. Thomassin, C. Jérôme and T. Tassaing, J. Supercrit. Fluids, 2014, 90, 44–52 CrossRef CAS.
- M. Takada, M. Tanigaki and M. Ohshima, Polym. Eng. Sci., 2001, 41, 1938–1946 CAS.
- Z. Zhong, S. Zheng and Y. Mi, Polymer, 1999, 40, 3829–3834 CrossRef CAS.
- D. Li, T. Liu, L. Zhao and W. Yuan, AIChE J., 2012, 58, 2512–2523 CrossRef CAS.
- M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1988, 1259–1260 RSC.
- N. Soultanidis, W. Zhou, A. C. Psarras, A. J. Gonzalez, E. F. Iliopoulou, C. J. Kiely, I. E. Wachs and M. S. Wong, J. Am. Chem. Soc., 2010, 132, 13462–13471 CrossRef CAS PubMed.
- T. K. Phung, L. Proietti Hernández and G. Busca, Appl. Catal., A, 2015, 489, 180–187 CrossRef CAS.
- D. Lopez, K. Suwannakarn, D. Bruce and J. Goodwinjr, J. Catal., 2007, 247, 43–50 CrossRef CAS.
- X.-F. Yu, N.-Z. Wu, H.-Z. Huang, Y.-C. Xie and Y.-Q. Tang, J. Mater. Chem., 2001, 11, 3337–3342 RSC.
- S. Eibl, B. C. Gates and H. Knözinger, Langmuir, 2001, 17, 107–115 CrossRef CAS.
- X. Chen, G. Clet, K. Thomas and M. Houalla, J. Catal., 2010, 273, 236–244 CrossRef CAS.
- T. Onfroy, V. Lebarbier, G. Clet and M. Houalla, J. Mol. Catal. A: Chem., 2010, 318, 1–7 CrossRef CAS.
- T. Onfroy, G. Clet and M. Houalla, J. Phys. Chem. B, 2005, 109, 3345–3354 CrossRef CAS PubMed.
- W. Zhou, E. I. Ross-Medgaarden, W. V. Knowles, M. S. Wong, I. E. Wachs and C. J. Kiely, Nat. Chem., 2009, 1, 722–728 CrossRef CAS PubMed.
- C. Hammond, J. Straus, M. Righettoni, S. E. Pratsinis and I. Hermans, ACS Catal., 2013, 3, 321–327 CrossRef CAS.
- K. Nishiwaki, N. Kakuta, A. Ueno and H. Nakabayashi, J. Catal., 1989, 118, 498–501 CrossRef CAS.
- L. F. Chen, L. E. Noreña, J. Navarrete and J. A. Wang, Mater. Chem. Phys., 2006, 97, 236–242 CrossRef CAS.
- J. B. Miller and E. I. Ko, J. Catal., 1996, 159, 58–68 CrossRef CAS.
- D. Lopez, J. Goodwinjr and D. Bruce, J. Catal., 2007, 245, 381–391 CrossRef CAS.
- A. S. Poyraz, C.-H. Kuo, E. Kim, Y. Meng, M. S. Seraji and S. L. Suib, Chem. Mater., 2014, 26, 2803–2813 CrossRef CAS.
- S. A. K. Leghari, S. Sajjad, F. Chen and J. Zhang, Chem. Eng. J., 2011, 166, 906–915 CrossRef CAS.
- R. T. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- L. Yang, Y. Xiao, S. Liu, Y. Li, Q. Cai, S. Luo and G. Zeng, Appl. Catal., B, 2010, 94, 142–149 CrossRef CAS.
- M. A. Cortés-Jácome, J. A. Toledo, C. Angeles-Chavez, M. Aguilar and J. A. Wang, J. Phys. Chem. B, 2005, 109, 22730–22739 CrossRef PubMed.
- F. Riboni, L. G. Bettini, D. W. Bahnemann and E. Selli, Catal. Today, 2013, 209, 28–34 CrossRef CAS.
- D.-S. Kim, J.-H. Yang, S. Balaji, H.-J. Cho, M.-K. Kim, D.-U. Kang, Y. Djaoued and Y.-U. Kwon, CrystEngComm, 2009, 11, 1621 RSC.
- A. Fuerte, M. D. Hernández-Alonso, A. J. Maira, A. Martínez-Arias, M. Fernández-García, J. C. Conesa, J. Soria and G. Munuera, J. Catal., 2002, 212, 1–9 CrossRef CAS.
- M. Fernández-García, A. Martínez-Arias, A. Fuerte and J. C. Conesa, J. Phys. Chem. B, 2005, 109, 6075–6083 CrossRef PubMed.
- N. B. Shali and S. Sugunan, Mater. Res. Bull., 2007, 42, 1777–1783 CrossRef CAS.
- R. Kourieh, V. Rakic, S. Bennici and A. Auroux, Catal. Commun., 2013, 30, 5–13 CrossRef CAS.
- H. K. Motonobu Goto, A. Kodama and T. Hirose, AIChE J., 2002, 48, 135 Search PubMed.
- C. D. Baertsch, K. T. Komala, Y.-H. Chua and E. Iglesia, J. Catal., 2002, 205, 44–57 CrossRef CAS.
- D. G. Barton, S. L. Soled, G. D. Meitzner, G. A. Fuentes and E. Iglesia, J. Catal., 1999, 181, 57–72 CrossRef CAS.
- C. D. Baertsch, S. L. Soled and E. Iglesia, J. Phys. Chem. B, 2001, 105, 1320–1330 CrossRef CAS.
- L. Vilcocq, P. C. Castilho, F. Carvalheiro and L. C. Duarte, ChemSusChem, 2014, 7, 1010–1019 CrossRef CAS PubMed.
- M. Zheng, J. Pang, A. Wang and T. Zhang, Chin. J. Catal., 2014, 35, 602–613 CrossRef CAS.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06298a |
| This journal is © The Royal Society of Chemistry 2016 |