
A photoactivatable Src homology 2 (SH2) domain†
X. Songa,
X. Shanga,
T. Jua,
R. Cernya,
W. Niu*b and
J. Guo*a
aDepartment of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588, USA. E-mail: jguo4@unl.edu
bDepartment of Chemical & Biomolecular Engineering, University of Nebraska-Lincoln, Lincoln, Nebraska 68588, USA. E-mail: wniu2@unl.edu
First published on 19th May 2016
Abstract
Src homology 2 (SH2) domains bind specifically to phosphotyrosine-containing motifs. As a regulatory module of intracellular signaling cascades, the SH2 domain plays important roles in the signal transduction of receptor tyrosine kinase pathways. In this work, we reported the construction of a photoactivatable SH2 domain through the combination of protein engineering and genetic incorporation of photo-caged unnatural amino acids. Significantly enhanced recognition of a phosphotyrosine-containing peptide substrate by the engineered SH2 domain mutant was observed after light-induced removal of the photo-caging group. Optical activation allows the control of protein and/or cellular function with temporal and spatial resolution. This photoactivatable SH2 domain could potentially be applied to the study of tyrosine phsophorylation-associated biological processes.
1. Introduction
Src homology 2 (SH2) domains represent the largest class of known phosphotyrosine (pTyr)-binding domains.1,2 A typical human cell is estimated to contain 120 non-redundant SH2 domains in 111 unique proteins, including kinases, phosphatases, cytoskeletal proteins, regulators of small GTPases, etc. The recognition of pTyr-containing proteins (phosphoproteins) or peptides (phosphopeptides) by SH2 domains is generally in a sequence-specific and phosphorylation-dependent manner. Phosphopeptides of optimal sequence bind to their cognate SH2 domains with dissociation constants in the range of 0.05–0.5 μM. Such specific interactions between SH2 domains and their phosphorylated partners are essential in the activation or the deactivation of protein functions and/or cellular processes.Here we report the construction of a photoactivatable SH2 domain through protein engineering and the genetic incorporation of photo-caged unnatural amino acid (Fig. 1 and 2). It represents the first example that the function of a SH2 domain can be turned on by light. Such conditional control of SH2 domain function would not only facilitate investigations of SH2 domain-associated cellular events, but also provide an optogenetic tool to regulate an intracellular signaling cascade of interest. Light activation strategy provides spatial and temporal resolution on the activation and/or deactivation of cellular function.
 | ||
| Fig. 1 A photoactivatable Src homology 2 (SH2) domain. The Src SH2 domain (PDB 1SPS) is shown in green. The phosphorylated peptide substrate is shown in yellow. The photo-caging group is shown as an orange circle. pTyr, phosphotyrosine. | ||
 | ||
| Fig. 2 Genetic incorporation of ONBK into GFPUV-Asn149 mutant. (A) The structure of ONBK; (B) GFPUV fluorescence assays. Fluorescence intensity was normalized to cell growth; (C) SDS-PAGE analysis of GFPUV-Asn149 expression. Lane M, molecular weight marker; lane 1, with ONBK; lane 2, without ONBK. ONBK, o-nitrobenzyl-oxycarbonyl-Nε-L-lysine. | ||
2. Experimental
2.1 Materials and general methods
Chemicals were purchased from Sigma or Fisher Scientific. Peptides were purchased from NeoBioLab, Inc. Primers were ordered from Sigma. Restriction enzymes, T4 DNA ligase, and Antarctic phosphatase (AP) were purchased from New England Biolabs. KOD hot start DNA polymerase was purchased from EMD Millipore. Standard molecular biology techniques3 were used throughout. DNA sequencing analyses were conducted by Eurofins Genomics. Site-directed mutagenesis was carried out using overlapping PCR. E. coli GeneHogs (Life Technologies) were used for routine cloning, DNA propagation, and protein expression. Plasmid preparation and DNA extraction kits were purchased from Zymo Research. All solutions were prepared in deionized water further treated by Barnstead Nanopure® ultrapure water purification system. Antibiotics were added where appropriate to following final concentrations: kanamycin, 50 mg L−1; chloramphenicol, 34 mg L−1. The synthesis procedure for ONBK can be found in ESI.†2.2 Screening of aminoacyl-tRNA synthetase
In a typical experiment, two tubes of E. coli GeneHogs cells harboring plasmid pLei-GFPUV-Asn149TAG and a pBK-PylRS variant of interest were grown in 5 mL LB media containing kanamycin and chloramphenicol at 37 °C. The protein expressions were induced at the OD600 of 0.6 by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG; 0.25 mM). ONBK was added to one of the two culture tubes to a final concentration of 1 mM. After an additional 16 h of cultivation, 1 mL of cell culture from each tube was collected, washed, and resuspended in 1 mL of potassium phosphate buffer (50 mM, pH 7.4). The two samples were subsequently used for fluorescence and cell density measurements using a Synergy™ H1 Hybrid plate reader (BioTek Instruments). The fluorescence of GFPUV was monitored at λEx = 390 nm and λEm = 510 nm. The cell density was estimated by measuring the sample absorbance at 600 nm. Values of fluorescence intensity were normalized to cell growth. Using the same set of experimental procedures, previously reported pyrrolysyl-tRNA synthetase (PylRS) mutants were screened for their ability to incorporate ONBK.2.3 Construction of plasmids
Four plasmids, pSH2-Arg35TAG, pSH2-Arg35Lys, pSH2-Arg35TAG-tm, and pSH2-Arg35Lys-tm were constructed. The SH2 domain-encoding gene was amplified from pET30b-Src4 using primer P1 and P4 (ESI†). The Arg35TAG mutation was introduced by overlapping PCR using primers P2 and P3 (ESI†). Digestion of the PCR product followed by ligation into the NdeI and XhoI sites of pLei vector5 afforded plasmid pSH2-Arg35TAG. Plasmid pSH2-Arg35Lys was constructed following the same procedure. The Arg35Lys mutation was introduced by overlapping PCR using primers P2 and P5 (ESI†). The introduction of Thr40Val, Cys45Ala, and Lys63Leu mutations was carried out by site-directed mutagenesis using primers P6–P9 (ESI†). All the SH2 domain variants were confirmed by DNA sequencing.2.4 Protein expression and purification
For the expression of SH2 domain that contains ONBK, E. coli GeneHogs cells harboring pBK-TocKRS and pSH2-Arg35TAG (or pSH2-Arg35TAG-tm) were cultured in 100 mL of LB medium with kanamycin and chloramphenicol at 37 °C. When the OD600 of the culture reached 1.0, the protein expression was induced by the addition of IPTG and ONBK to a final concentration of 0.25 mM and 1 mM, respectively. After 24 h of cultivation at room temperature, cells were collected by centrifugation (5 000g, 10 min), resuspended in lysis buffer (20 mM potassium phosphate, pH 7.4, 150 mM NaCl, and 20 mM imidazole), and disrupted by sonication. Cellular debris was removed by centrifugation (21 000g, 30 min, 4 °C). The cell-free lysate was applied to Ni Sepharose 6 Fast Flow resin (GE Healthcare). Protein purification followed manufacture's instructions. Purified protein was eluted off the Amicon Econo-Pac® 10DG desalting column (BioRad Laboratories, Inc) using storage buffer that contains Tris–HCl (25 mM, pH 7.5), NaCl (150 mM), and glycerol (5%).The same procedure was applied to the expression of SH2 domains containing the Arg35Lys mutation (SH2-Arg35Lys and SH2-Arg35Lys-tm) with the exception that plasmid pBK-TocKRS was not included in the protein expression strains and ONBK was not added to the culture media.
2.5 De-caging
An aliquot of SH2 domain protein (22.2 μM) was irradiated by handheld UV illuminator for 0 min, 15 min, 30 min, or 60 min at 365 nm (or 302 nm). During the irradiation, the protein samples were chilled on ice to avoid extensive heating from UV light.2.6 Fluorescence polarization (FP) assay
The fluorescein-labeled peptide probe was dissolved in assay buffer containing potassium phosphate (20 mM, pH 7.35), NaCl (100 mM), DTT (2 mM), and bovine gamma globulin (0.1%). Aliquot of the peptide solution was distributed in 96-well fluorescence plate (Corning Costar 3915) to reach a final concentration of 10 nM. After the addition of indicated amounts of a SH2 domain variant of interest (20 μM), the assay solution was gently mixed and incubated at room temperature in the dark for 25 minutes. Fluorescence polarization measurements were performed on a Synergy™ H1 hybrid plate reader (BioTek Instruments, Inc) equipped with filter cube (λEx = 485 nm, BP = 20 nm; λEm = 528 nm, BP = 20 nm). A standard sample layout in 96-well plate was designed so that a single plate contains all the samples for one SH2 domain variant. Triplet samples of each protein concentration were arrayed in the plate, together with control samples containing either only the SH2 mutant protein or peptide probe. Fluorescence intensity data were exported, converted into anisotropy values. Calculation of the percentage of bound probe followed reported method.6 Dissociation constants were calculated by curve fitting into one-site specific binding equation with Hill slope using GraphPad Prism 5 (GraphPad Software, Inc). Fluorescence polarization assays of all SH2 domain variants were repeated three times. Results were reported as the average with standard deviation.3. Results and discussion
3.1 General approach and rationale
In order to control the affinity between an SH2 domain and its binding partner, key interaction(s) between the two entities need(s) to be manipulated. It is known that the pTyr residue makes a major contribution to the binding free energy (ΔG°) of a phosphopeptide to its cognate SH2 domain.7 We also observed that the dissociation constant between the Src SH2 domain (from the proto-oncogene tyrosine-protein kinase Src) and its peptide substrate was highly dependent on the phosphorylation state of a tyrosine residue. With phosphorylation of the tyrosine, the KD value was in the range of 0.061 ± 0.008 μM.4 In contrast, the actual KD value (>15 μM) was too large to be accurately measured when the peptide substrate was not phosphorylated. Indeed, electrostatic forces between the two positively charged Arginine (Arg) residues of the SH2 domain (e.g., Arg15 and Arg35 in the Src SH2 domain)8,9 and the negatively charged pTyr residue of the peptide substrate are the key interactions. Besides the pTyr residue, the two Arg residues, which are highly conserved in all known human SH2 sequences,10 do not have any apparent interactions with other part of the peptide substrate.8,9 We hypothesized that the interaction between SH2 domain and its binding partner could be largely abolished if the two Arg residues are masked with uncharged photo-caging groups. After light-induced cleavage of the caging group, the restoration of the two Arg residues would yield a functional SH2 domain (Fig. 1).Since the simultaneous introduction of two bulky photo-caging groups may impose significant structural distortion of an SH2 domain, we decided to mask only one of the two Arg residues. We envisaged that the loss of a single Arg residue would still considerably weaken the recognition of pTyr by the SH2 domain. In fact, we demonstrated that high affinity binding of pTyr by Src SH2 domain required the presence of both Arg15 and Arg35 residues. According to our measurements, the Arg15Gln mutant and the Arg35Gln mutant displayed nearly 40-fold lower affinity towards a phosphopeptide probe (Fig. 3A) than that of the wild-type SH2 domain. In addition to the loss of a positive charge, the installation of a caged-Arg may further impair the function of SH2 domain due to the bulkiness of the caging group.
 | ||
| Fig. 3 Recognition of phosphopeptide by the SH2-Arg35ONBK mutant with and without UV irradiation. (A) The structure of the phosphopeptide. (B) Fluorescence polarization assay. Each data point is the average of triplicate measurements with standard deviation. | ||
As the genetic incorporation of photo-caged Arg has not been developed, we decided to use a photo-caged lysine (Lys) instead. Both Arg and Lys are positively charged amino acids. While the side chain of Lys is shorter than that of Arg, it is possible that the replacement of an Arg residue with a Lys residue may not significantly affect the function of a SH2 domain. Indeed, the SH2-Arg35Lys mutant showed a similar affinity (KD = 0.117 μM) to that (KD = 0.061 μM) of the wild-type SH2 domain (SH2-wt). On the other hand, mutation at position Arg15 afforded a less active SH2 domain variant (unpublished data). Therefore, we focused on the engineering of the SH2-Arg35Lys mutant in this work. Unmasking of a photo-caged lysine at position 35 would afford a functional SH2 domain.
3.2 Genetic incorporation of a photo-caged lysine
A previously reported photo-caged lysine, o-nitrobenzyl-oxycarbonyl-Nε-L-lysine (ONBK; Fig. 2A),11 was employed in this work. The ortho-nitrobenzyl group represents one of the most frequently used photo-caging groups in biological experiments. It is chemically inert in living cells and has a good quantum yield of de-caging within a short timescale. Furthermore, the de-caging of the ortho-nitrobenzyl group can be achieved using a near visible UV light (e.g., 365 nm), which is generally considered as safe to proteins and cells.Genetic incorporation of ONBK into proteins in response to amber nonsense codon in E. coli was reported.11 In order to replace Arg35 with ONBK, the codon of Arg35 was mutated into an amber nonsense codon. By using the evolved ONBK-specific pyrrolysyl-tRNA synthetase mutant (NBK-1)11 together with its cognate amber suppressor tRNA (tRNACUA), the desired SH2 domain mutant, SH2-Arg35ONBK, was obtained. However, the protein yield (less than 0.2 mg L−1) was not to our satisfaction. While a series of different cultivation conditions were examined, no significant improvement in protein yield could be obtained. We concluded that the activity of NBK-1 was the likely limiting factor in the expression of SH2-Arg35ONBK.
In order to improve the expression level of SH2-Arg35ONBK, we subsequently screened a library of previously evolved pyrrolysyl-tRNA synthetase (PylRS) variants. A fluorescence-based screening assay was used to gauge the catalytic activity of PylRS variants in the aminoacylation of tRNACUA with ONBK. The amber suppression efficiency was directly linked to the expression level of a GFPUV mutant (GFPUV-Asn149TAG)11 that contains an amber nonsense codon at position Asn149. Among all the PylRS variants examined (Fig. S1, ESI†), DizPKRS,12 BhcKRS,13 and TcokRS,14 showed excellent efficiency in the incorporation of ONBK into GFPUV-Asn149 mutant protein in response to amber nonsense codon (Fig. 2B). Based on the fluorescence assay (Fig. S1, ESI†), the DizPKRS, BhcKRS, and TcokRS mutants displayed about 68, 100, and 116 folds higher efficiency in ONBK incorporation than that of the reported NBK-1 mutant, respectively. Furthermore, notable GFP fluorescence was only observed in the presence of ONBK for all three PylRS variants (Fig. 2B). This result indicates that these PylRS variants only charge tRNACUA with ONBK but not with any of the endogenous natural amino acids in E. coli. Among the three hits, TcokRS showed the best fidelity and efficiency towards ONBK. Subsequent SDS-PAGE analysis (Fig. 2C) confirmed the conclusion of the fluorescence-based assay. Therefore, TcokRS was selected for the genetic incorporation of ONBK into SH2 domain mutants in this work.
3.3 The first generation SH2 domain mutant
The expression of the SH2-Arg35ONBK mutant (encoded by plasmid pSH2-Arg35TAG) was carried out in LB medium in the presence of tRNACUA and TcokRS, supplemented with 1 mM ONBK. The estimated protein yield was over 6 mg L−1 following partial purification by affinity chromatography. The tandem mass spectrometry data (Fig. S2, ESI†) confirmed the site-specific incorporation of ONBK.The affinity between the SH2-Arg35ONBK mutant and a fluorescein-labeled peptide probe was measured by fluorescence polarization assay (Fig. 3). The peptide probe has the exact amino acid sequence of the natural substrate of Src SH2 domain. The fluorescein was conjugated to the peptide substrate through a N-terminal glycine. As shown in Fig. 3B, the caged SH2-Arg35ONBK bound to the peptide probe with very low affinity (KD = 4.972 ± 0.436 μM). This result was consistent with our hypothesis that a single Arg35 to ONBK mutation could significantly reduce the ability of SH2 domain to recognize pTyr. After 15 minutes of UV (365 nm) irradiation, the SH2-Arg35ONBK mutant displayed about 4-fold higher affinity towards the peptide probe (KD = 1.273 ± 0.533 μM; Fig. 3B). The apparent KD values continued to decrease as the time of irradiation increased (Fig. 3B). A plateau (KD = 0.690 ± 0.053 μM) was reached following 30 minutes of UV irradiation. Longer irradiation time did not led to further increase in binding affinity.
While the initial results showed that the function of the SH2-Arg35ONBK mutant could be partially turned on by UV irradiation, only 7.4-fold affinity enhancement was obtained. In comparison to SH2-Arg35Lys (KD = 0.117 μM), the UV-irradiated SH2-Arg35ONBK displayed a significantly lower affinity (KD = 0.669 ± 0.053 μM). To rule out potential damages of SH2 domain function caused by UV irradiation, the affinity between SH2-Arg35Lys and the peptide probe was examined with and without UV irradiation. No detectable effect by UV irradiation was observed (Fig. S3, ESI†). Therefore, we hypothesized that the low de-caging efficiency of ONBK was the limiting factor in the restoration of SH2 domain function.
3.4 The second generation SH2 domain mutant
According to literature, a clean de-caging of ONBK that locates on the surface of a GFP protein could be achieved within 20 minutes of UV irradiation.11 We hypothesized that a crowded binding pocket containing chemically reactive amino acid side chains could interfere with the de-caging reaction. In order to improve the de-caging efficiency, we further engineered the pTyr-binding pocket of the SH2-Arg35ONBK protein by introducing three mutations (Thr40Val, Cys45Ala, and Lys63Leu) to afford SH2-Arg35ONBK-tm (Fig. 4). These mutations partially opened up the binding pocket (Fig. 4) by substitution of amino acids with side chains of relatively smaller sizes. In the meantime, three chemically reactive amino acid side chains (a secondary alcohol, a thiol, and a primary amine) were removed. | ||
| Fig. 4 Engineering of SH2 domain. | ||
As shown in Fig. 5, the SH2-Arg35ONBK-tm protein showed significantly lower affinity (KD > 15 μM) towards the peptide substrate than that of SH2-Arg35ONBK (KD = 4.972 ± 0.436 μM; Fig. 3). It should be noted that the interaction between SH2-Arg35ONBK-tm and the phosphopeptide was too weak to be accurately measured. The fluorescence intensity values could not reach a clear plateau even in the presence of very high concentrations of protein. On the other hand, after 60 minutes of UV irradiation, the SH2-Arg35ONBK-tm protein displayed significantly higher affinity (KD = 0.295 ± 0.022 μM; Fig. 5) than that of the irradiated SH2-Arg35ONBK. An at least 50-fold change in affinity was observed before and after UV irradiation. Introduction of the three mutations (Thr40Val, Cys45Ala, and Lys63Leu) into SH2-Arg35ONBK therefore lowered the background binding and increased signal output after the UV irradiation. We attribute these improved properties of SH2-Arg35ONBK-tm to following possible factors: (1) the Lys63Leu mutation eliminates the electrostatic interaction between Lys63 in SH2-Arg35ONBK and a glutamate residue on the peptide substrate (Fig. 3A and 4), which reduces the non-pTyr-dependent interaction and leads to lower initial affinity of the caged SH2 protein; (2) the Thr40Val, Cys45Ala, and Lys63Leu mutations improve the affinity of de-caged SH2-Arg35ONBK-tm towards phosphopeptide through the formation of a hydrophobic surface that engages the phenyl ring of the pTyr residue;15 and (3) the bulky caging group efficiently prevents the optimal interaction between the hydrophobic surface in caged SH2-Arg35ONBK-tm and the phenyl ring of the pTyr residue in phosphopeptide. The apparently additive effects of these factors are translated into an excellent dynamic range in binding affinities that are controlled by UV irradiation.
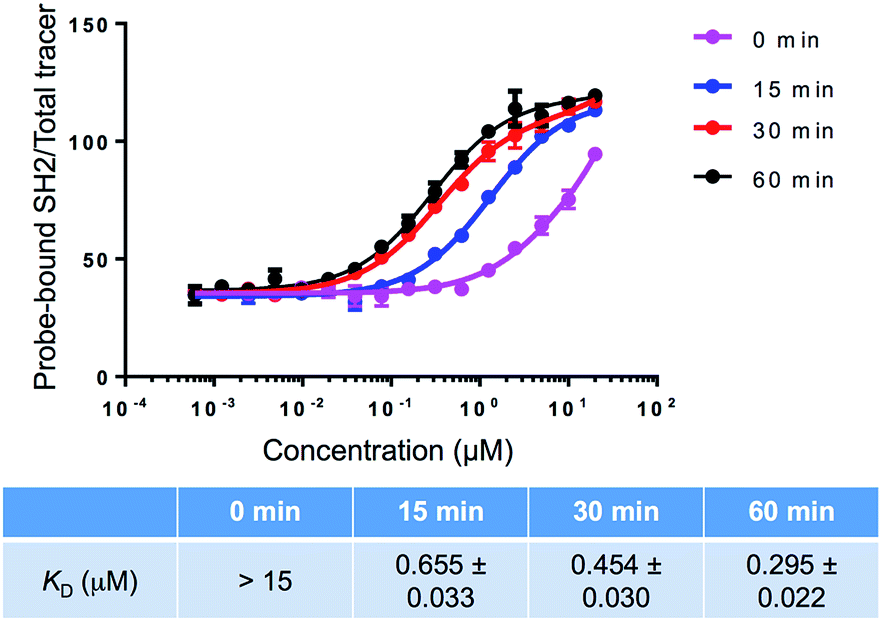 | ||
| Fig. 5 Recognition of phosphopeptide by the SH2-Arg35ONBK-tm mutant with and without UV irradiation. Each data point is the average of triplicate measurements with standard deviation. | ||
4. Conclusions
In summary, we have constructed the first photoactivatable SH2 domain mutant, which can be potentially applied as a general optogenetic tool to the photocontrol of pTyr-associated biological processes. The protein engineering approach enabled us to use a photo-caged lysine to mask and unmask the function of a key arginine residue of SH2 domain. Additional engineering efforts will be made towards further improving the property of the obtained photoactivatable SH2 domain mutant. Other photo-caging groups will also be examined. The approaches and findings from this work are likely applicable to the engineering of other photoactivatable SH2 domains.Acknowledgements
This work is supported by the New Faculty Startup Fund to J. G. from the Chemistry Department of University of Nebraska – Lincoln and by Grant CBET1264708 (to J. G. and W. N.) from National Science Foundation.References
- B. J. Mayer, P. K. Jackson and D. Baltimore, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 627–631 CrossRef CAS.
- B. A. Liu, K. Jablonowski, E. E. Shah, B. W. Engelmann, R. B. Jones and P. D. Nash, Mol. Cell. Proteomics, 2010, 9, 2391–2404 CAS.
- Molecular cloning: A laboratory manual, ed. J. F. Sambrook and D. W. Russell, Cold Spring Harbor Laboratory Press, 3rd edn, 2000 Search PubMed.
- T. Ju, W. Niu, R. Cerny, J. Bollman, A. Roy and J. Guo, Mol. BioSyst., 2013, 9, 1829–1832 RSC.
- W. Niu, P. G. Schultz and J. Guo, ACS Chem. Biol., 2013, 8, 1640–1645 CrossRef CAS PubMed.
- A. M. Rossi and C. W. Taylor, Nat. Protoc., 2011, 6, 365–387 CrossRef CAS PubMed.
- G. Waksman, S. Kumaran and O. Lubman, Expert Rev. Mol. Med., 2004, 6, 1–18 Search PubMed.
- G. Waksman, S. E. Shoelson, N. Pant, D. Cowburn and J. Kuriyan, Cell, 1993, 72, 779–790 CrossRef CAS PubMed.
- G. Waksman, D. Kominos, S. C. Robertson, N. Pant, D. Baltimore, R. B. Birge, D. Cowburn, H. Hanafusa and B. J. Mayer, Nature, 1992, 358, 646–653 CrossRef CAS PubMed.
- SH2 domain website: by the Nash Lab, http://sites.google.com/site/sh2domain/alignment, accessed Jan 09.
- P. R. Chen, D. Groff, J. Guo, W. Ou, S. Cellitti, B. H. Geierstanger and P. G. Schultz, Angew. Chem., Int. Ed., 2009, 48, 4052–4055 CrossRef CAS PubMed.
- M. Zhang, S.-X. Lin, X.-W. Song, J. Liu, Y. Fu, X. Ge, X.-M. Fu, Z.-Y. Chang and P.-R. Chen, Nat. Chem. Biol., 2011, 7, 671–677 CrossRef CAS PubMed.
- J. Luo, R. Uprety, Y. Naro, C. Chou, D. P. Nguyen, J. W. Chin and A. Deiters, J. Am. Chem. Soc., 2014, 136, 15551–15558 CrossRef CAS PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- T. Kaneko, H. Huang, X. Cao, X. Li, C. Li, C. Voss, S. S. Sidhu and S. S. C. Li, Sci. Signaling, 2012, 5, ra68 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06211c |
| This journal is © The Royal Society of Chemistry 2016 |