
Phosphorescence of free base corroles†
Abstract
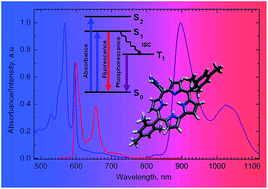
The phosphorescence spectra of a series of free base halogenated meso-triarylcorroles are obtained and systematically analysed for the first time. Phosphorescence quantum yields and lifetimes are reported and rationalised in terms of the internal heavy atom effects, and the reasons for the unusually large S1–T1 energy gap in the studied compounds are discussed.
 Please wait while we load your content...
Please wait while we load your content...

