
Factors affecting oxygen evolution through water oxidation on polycrystalline titanium dioxide†
Yuuya Nishimotoa,
Yuichi Hasegawab,
Kenta Adachia and
Suzuko Yamazaki*a
aDivision of Environmental Science and Engineering, Graduate School of Science and Engineering, Yamaguchi University, Yamaguchi 753-8512, Japan. E-mail: yamazaki@yamaguchi-u.ac.jp
bDepartment of Biology and Chemistry, Faculty of Science, Yamaguchi University, Yamaguchi 753-8512, Japan
First published on 26th April 2016
Abstract
The effect of physicochemical properties such as specific surface area, crystalline phase, crystallite size, and crystallinity of TiO2 on photocatalytic water oxidation was investigated. Two types of polycrystalline TiO2 samples which have different physicochemical properties were prepared by a sol–gel method. The photocatalytic activities of the TiO2 samples for water oxidation were evaluated by the O2 evolution rate from an aqueous silver nitrate solution under ultraviolet light irradiation. Comparison of the two types of TiO2 samples revealed that the specific surface area and crystalline phase were not related to the O2 evolution rate. In contrast, a linear relationship between the crystallite sizes and the O2 evolution rates was observed for most of the TiO2 samples. Crystal face selective Pt and PbO2 depositions on the TiO2 samples were observed by photocatalytic reduction of PtCl62− and oxidation of Pb2+ ions, respectively, indicating that the oxidation and reduction sites are separated on the surface. An increase in the crystallite size of the TiO2 promotes a spatial separation of the redox sites and suppresses the electron–hole recombination, leading to the enhancement of the photocatalytic activity.
Introduction
Development of new energy sources has been required for the solution of energy problems. Since Fujishima and Honda1 reported photoelectrochemical water splitting using a single-crystal rutile TiO2 electrode in 1972, H2 and O2 production by photocatalytic water splitting has been widely studied as a promising technology for sustainable energy systems. Inorganic semiconductor materials such as TiO2,2–5 SrTiO3,6–8 and WO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9–11 are most promising photocatalysts for water splitting. Tantalates and metal oxides with d10 electric configuration have been widely studied for practical application in photocatalytic water splitting.12–17 However, at this stage, a photocatalyst having sufficient activity for practical use has not been developed. Thus, improvement of water splitting activity of inorganic semiconductor photocatalysts is strongly desired.
9–11 are most promising photocatalysts for water splitting. Tantalates and metal oxides with d10 electric configuration have been widely studied for practical application in photocatalytic water splitting.12–17 However, at this stage, a photocatalyst having sufficient activity for practical use has not been developed. Thus, improvement of water splitting activity of inorganic semiconductor photocatalysts is strongly desired.
Water splitting reaction consists of two half reactions: H2 evolution by reduction of protons and O2 evolution by water oxidation. The latter is the rate-limiting step in the overall reaction.18 Therefore, the improvement of O2 evolution is important to achieve high-efficiency water splitting. The O2 evolution rate depends on the calcination temperature of the photocatalysts due to a change in some physicochemical properties. Kominami et al.19 reported an enhancement of the O2 evolution on TiO2 calcined at higher temperature in the range of 250–1273 K and attributed it to the crystallization from amorphous to anatase and rutile. Recently, Maeda et al.20 described the enhancement of O2 evolution by calcining the commercially available rutile TiO2 at 873–1273 K in air, which can be ascribed to the improvement of charge separation due to the increase in particle size. Thalluri et al.21 reported the relationships between the O2 evolution rate on monoclinic BiVO4 and the physicochemical properties which were changed by the calcination temperature. Increase in the crystallite size and decrease in the band gap and the V–O bond length depending on the calcination temperature led to the enhancement of O2 evolution on BiVO4. Large particle size or crystallite size appears to be important for inorganic semiconductor photocatalysts to improve O2 evolution activity, although the reason has not been fully elucidated.
Other factors may also affect O2 evolution activity. High specific surface area is believed to be important for high photocatalytic activity.22–25 On the other hand, a small effect of specific surface area on water splitting activity also has been suggested.19,21,26,27 Dependence of photocatalytic activity on crystalline phases has been reported for some inorganic semiconductor photocatalysts. Monoclinic BiVO4 shows higher activity for O2 evolution than tetragonal BiVO4.28,29 Superior activity of rutile TiO2 for O2 evolution than anatase TiO2 has been reported,30,31 whereas anatase TiO2 shows higher activity for the decomposition of organic compounds than rutile TiO2.32–34 However, Prieto-Mahaney et al.35 have reported that the O2 evolution rate on TiO2 is not affected by the crystalline phases and convincing evidence for the influence of crystalline phases of TiO2 on O2 evolution has not been obtained. Degree of crystallinity of inorganic semiconductor photocatalysts is also considered to affect O2 evolution activity. Kho et al.36 have reported an enhancement of O2 evolution rate on BiVO4 with tetragonal and/or monoclinic phases by increasing calcination temperature and attributed it to the increase in crystallinity and monoclinic content. Ohtani et al.37 have investigated the photocatalytic activity of amorphous and anatase mixed TiO2 and have shown that an increase in the fraction of anatase leads to an enhancement of the photocatalytic activity.
The fact that the calcination of inorganic semiconductor photocatalysts leads to the enhancement of their O2 evolution activity should give us a clue for the development of highly active water splitting photocatalysts. However, the physicochemical properties of inorganic semiconductor photocatalysts change simultaneously depending on calcination temperature and thus the contribution of each physicochemical property to O2 evolution activity is still unclear. In this study, we focused on TiO2 which is the most typical inorganic semiconductor photocatalyst. Two types of TiO2 samples with different physicochemical properties were prepared by unique synthetic procedures. Comparison of their O2 evolution activity allowed the direct evaluation of the relationships between the physicochemical properties and the photocatalytic activity for water oxidation.
Experimental section
Materials
Titanium(IV) tetraisopropoxide (Ti(OC3H7)4) and dialytic membranes with 3500 molecular weight cut-off were purchased from Wako Pure Chemical Industries Ltd (Osaka, Japan) and Spectrum Laboratories (CA, USA), respectively. Ultrapure water with a specific resistivity of 18.2 MΩ cm was obtained using a Milli-Q water purification system (Millipore, USA) and used for all experiments. All other chemicals were of reagent grade, purchased from Wako Pure Chemical Industries Ltd and used without further purification.Preparation of TiO2 powders
Preparation of the TiO2 samples was as described previously.38 Approximately 15 mL of Ti(OC3H7)4 was dropped in 180 mL of aqueous solutions containing HNO3 (1.3 mL, 60%). The mixture was peptized at room temperature for 6 days to form a highly dispersed colloidal solution. This TiO2 sol was put in dialytic membrane pipes, dialyzed in 3000 mL of water for 3 days until a pH of approximately 4 was obtained and then dried at 40 °C for 3 days. The obtained xerogel was crushed into powder by using a mortar and pestle. The resulting powder was denoted as TiO2-D. The TiO2 powder which was denoted as TiO2-ND was prepared similarly without conducting dialysis. The TiO2-D and TiO2-ND powders were calcined at 200–900 °C for 2–10 hours with a ramping rate of 3 °C min−1. Unless otherwise stated, calcination for 2 hours is the standard in this paper.Characterization techniques
Nitrogen adsorption–desorption isotherm measurements were performed at −195.8 °C with a TriStar II 3020 analyzer (Micromeritics, USA). The specific surface areas were measured using the Brunauer–Emmett–Teller (BET) method. By using the Barrett–Joyner–Halenda (BJH) model, the pore size distributions were derived from the adsorption branches of isotherms. X-ray diffraction (XRD) analysis was performed using a MiniFlex600 (Rigaku, Japan) with Cu Kα radiation (40 kV, 15 mA) at 2θ angles from 10° to 90° with a scan speed of 10° min−1. The crystallite sizes of the TiO2 samples were estimated using the Scherrer formula:
 | (1) |
Scanning electron microscopy (SEM) images of the TiO2 samples were obtained with a JSM-7600F instrument (JEOL, Japan). For SEM measurements, the as-prepared TiO2 samples were attached onto the sample stage using double-sided carbon tapes. Then the attached samples on the stage were coated with platinum by spattering to prevent charging.
Transmission electron microscopy (TEM) images of the TiO2 samples were obtained with a JEM-2100 instrument (JEOL, Japan). Before TEM measurements, the TiO2 samples were dispersed in ethanol by sonication. The TEM samples were prepared by dropping of the TiO2 suspension onto a copper grid covered with a carbon film and followed by drying in air.
The degrees of crystallinities of the TiO2 samples were calculated by the following formula:
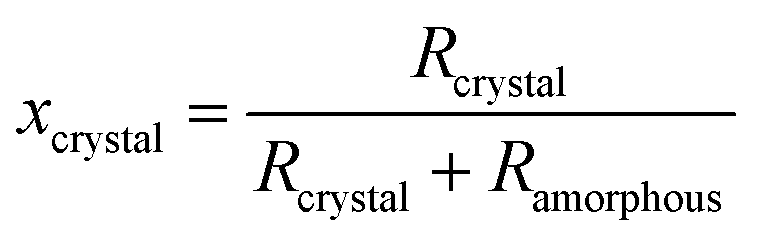 | (2) |
Evaluation of O2 evolution activity
Photocatalytic activity was evaluated by the O2 evolution rate from an aqueous AgNO3 solution which acts as an electron scavenger (eqn (3)):| Ag+ + e− → Ag | (3) |
A TiO2 sample (0.3 g) was suspended in 150 mL of AgNO3 solution (0.05 mol L−1) in a Pyrex reactor and then the reactor was immersed in a water bath thermostated at 30 °C on a magnetic stirrer. After air dissolved in the reactant solution was removed by bubbling argon gas for 30 min, the reactor was capped with a Mininert valve. Ultraviolet light (UV) irradiation (Fig. S1†) was provided by a 250 W super-high-pressure Hg lamp (Ushio Inc., Japan) using a U330 bandpass filter (HOYA CANDEO OPTRONICS, Japan). The amount of the evolved O2 was measured with a GC-8A gas chromatograph (Shimadzu, Japan, TCD, MS-5 Å column, Ar carrier).
Photodeposition of Pt and PbO2 on the TiO2 samples
In reference to previous reports,39,40 photodeposition of platinum (Pt) on the surface of the TiO2-ND calcined at 800 °C for 2 hours was performed using hexachloroplatinic(IV) acid (H2[PtCl6]) and ethanol, where Pt4+ was reduced to metal Pt by photogenerated electrons and ethanol was oxidized by photogenerated holes. 0.1 g of the TiO2-ND sample was suspended in 60 mL of 50 vol% aqueous ethanol solution. After addition of aqueous H2[PtCl6] solution (1000 mg L−1, 1 mL), dissolved O2 was removed by Ar purging. Subsequently, the suspension was irradiated with UV light under Ar purging for 3 hours. The Pt-loaded TiO2 sample was collected by filtration, washed several times with water and dried in an oven at 40 °C for 1 day. The obtained Pt-loaded TiO2 sample was suspended in 0.1 mol L−1 aqueous lead(II) nitrate (Pb(NO3)2) solution (60 mL) and pH of the suspension was adjusted to 1.0 with HNO3. UV light irradiation of the suspension and collection of the TiO2 sample co-loaded with Pt and PbO2 were carried out by the same procedure as for the Pt loading, although air was used as purge gas instead of Ar to provide O2.Results and discussion
Characterization of TiO2 samples
The specific surface areas of TiO2-D and TiO2-ND calcined at 200–600 °C for 2 hours are shown in Fig. 1. When being calcined at less than 500 °C, the specific surface areas of TiO2-D were higher than those of TiO2-ND. In the temperature range of 600–900 °C, only slight differences in the specific surface areas were observed. As shown in Fig. S2,† the pore size distribution of TiO2-D indicated the presence of mesopores with a peak at ca. 3 nm when being calcined at 200 °C, and such pore structures collapsed gradually with an increase in the calcination temperature. Differential values of dV/dD (V: pore volume; D: pore diameter) for TiO2-ND were lower than those for TiO2-D, indicating that the latter is a more porous material than the former. This fact is responsible for the higher specific surface area for TiO2-D than TiO2-ND. For the synthesis of TiO2-D, the pH value of the TiO2 sol increased from <1 to ca. 4 during the dialysis and the surface charges on the TiO2 particles decreased gradually on approaching its isoelectric point of ca. pH 6 (Fig. S3†). Such a decrease in the net surface charges would weaken electrostatic repulsion between the TiO2 particles to form aggregates with mesopores. | ||
| Fig. 1 Specific surface areas of TiO2-D and TiO2-ND calcined at 200–600 °C for 2 hours. | ||
The crystalline phase and crystallite size of TiO2-D and TiO2-ND were determined by powder XRD (Fig. S4 and S5†). When being calcined at 200 °C, both TiO2-D and TiO2-ND are anatase (JCPDS no. 21-1272) and there are no peaks ascribed to rutile. A very small diffraction peak at 2θ = 30.7° attributable to brookite (121) plane (JCPDS no. 29-1360) was observed for TiO2-D and TiO2-ND calcined at 200–400 °C. With an increase in the calcination temperature, a small diffraction peak at 2θ = 27.4° corresponding to rutile (110) plane (JCPDS no. 21-1276) first appeared for TiO2-D and TiO2-ND which were calcined at 400 °C and 300 °C, respectively. The phase transition from anatase to rutile is completed at 600 °C for TiO2-D or 800 °C for TiO2-ND. Fig. 2(a and b) shows the crystal phase compositions of the TiO2-D and TiO2-ND samples calcined at 200–900 °C. Zhang et al.41 reported that rutile nucleates at interfaces of the contacting anatase grains and thus the phase transformations from anatase to rutile take place at lower temperatures for smaller particles (<10 nm). On the basis of this interfacial nucleation mechanism, the densely aggregated TiO2-ND should transform to rutile at lower calcination temperature than TiO2-D which has lots of mesopores.42–44 Therefore, the lower transformation temperature from anatase to rutile observed for TiO2-D cannot be explained in terms of the interfacial nucleation mechanism. Inagaki et al.45 have reported that mixing of polyvinyl alcohol with TiO2 delayed the phase transition from anatase to rutile compared with pure TiO2. To investigate the presence of organic matter, TG and DTA measurements for TiO2-D and TiO2-ND calcined at 200 °C were performed (Fig. S6†). For both TiO2-D and TiO2-ND, large mass reduction was observed in the temperature range up to 400 °C and attributed to the evaporation of moisture and the decomposition of organic components.46 The mass reduction of TiO2-D or TiO2-ND was estimated to be 10.8% or 14.9%, respectively. This finding indicates that more organic residue exists in TiO2-ND prepared without dialysis, which causes the slower phase transition from anatase to rutile. On the other hand, the phase transformation behaviors from brookite to rutile in TiO2-D and TiO2-ND are similar. The mass fractions of brookite in TiO2-D and TiO2-ND calcined at 200–400 °C are maintained at around 30% as shown in Fig. 2(a and b). The phase transformations from brookite to rutile within both TiO2-D and TiO2-ND are almost completed by calcination at 500 °C. It is noted that for TiO2-ND calcined at 300–500 °C, no significant change in the mass fraction of anatase is observed. These results indicate that the phase transformations from brookite to rutile in TiO2-D and TiO2-ND proceed directly. Li et al.47 have also reported that brookite transforms to rutile directly and the phase transformation mainly proceeds within the individual TiO2 particles. Fig. 2(c and d) shows the crystallite sizes of the TiO2-D and TiO2-ND samples. The crystallite sizes of all crystalline phases in TiO2-D and TiO2-ND are increased with an increase in the calcination temperature.
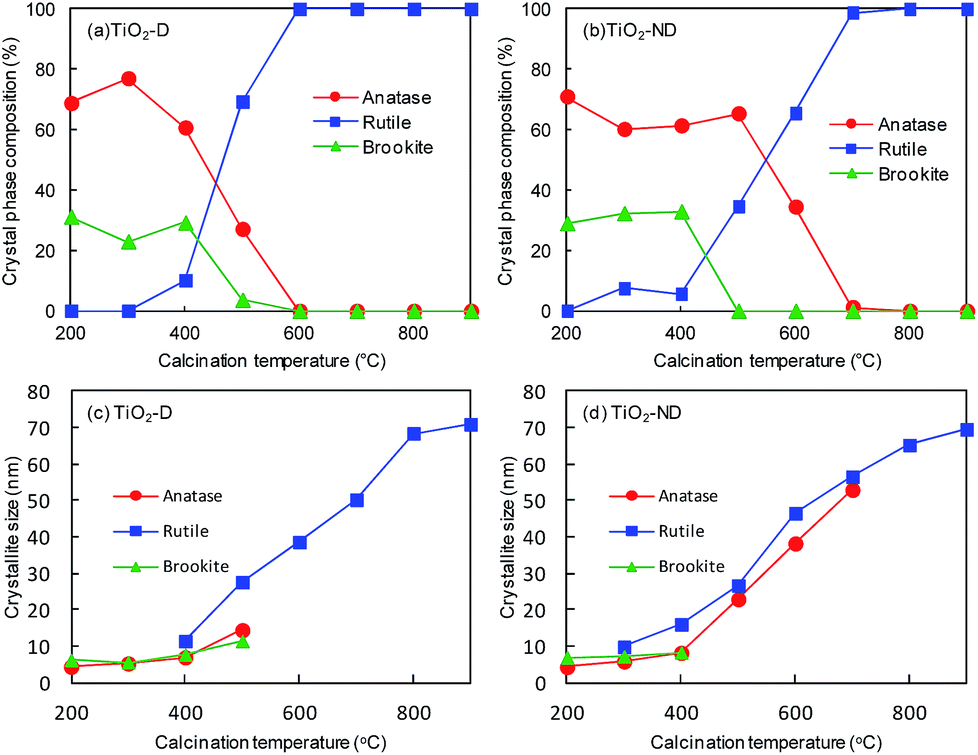 | ||
| Fig. 2 (a, b) Crystalline phase compositions and (c, d) crystallite sizes of TiO2-D and TiO2-ND calcined at 200–900 °C for 2 hours. | ||
Fig. 3(a) shows the mass fraction of the crystalline, amorphous, and impurity components in TiO2-D calcined at 200–900 °C. The mass fraction of impurities is reduced with increasing calcination temperature. This result is attributable to combustion of organic matters by the calcination and reduction in the amount of adsorbed water due to a decrease in the specific surface area. Surprisingly, the mass fractions of crystalline and amorphous components hardly change on calcination. The degree of crystallinity of TiO2-D calculated by eqn (2) is shown in Fig. 3(b) and significant change depending on the calcination temperature is not observed. In order to confirm the accuracy of this evaluation method, the crystallinity of a commercially available TiO2 (TiO2-Wako-rutile, rutile form, 99.9%, Wako Pure Chemical Industries Ltd) was also investigated and estimated to be 97.7%. This result guarantees the high accuracy of the method.
 | ||
| Fig. 3 (a) Component composition and (b) degree of crystallinity of TiO2-D calcined at 200–900 °C for 2 hours. | ||
O2 evolution activities of TiO2 samples
Fig. 4 shows the time courses of O2 evolution on TiO2-D and TiO2-ND calcined at 200–900 °C. The O2 evolution rates on TiO2-D and TiO2-ND calcined at the same temperature are very similar. As mentioned above, the specific surface areas of TiO2-D calcined at 200–500 °C are significantly higher than those of TiO2-ND calcined at the same temperature. These results clearly indicate that O2 evolution activity of TiO2 is not affected by the specific surface area. Furthermore, the O2 evolution rate on TiO2 hardly depends on the Ag+ concentration (0.001–0.01 mol L−1) and the O2 evolutions are suddenly stopped by the depletion of Ag+ (Fig. S7†). This result indicates that Ag+ is easily reduced on TiO2 and the decrease in the specific surface area by the deposition of Ag hardly affects the O2 evolution rate. | ||
| Fig. 4 Time courses of O2 evolution from a 0.05 mol L−1 AgNO3 solution under UV light irradiation. TiO2-D and TiO2-ND samples were calcined at (a) 200, (b) 300, (c) 400, (d) 500, (e) 600, (f) 700, (g) 800, and (h) 900 °C for 2 hours. | ||
As shown in Fig. 2(a and b), the crystalline phase compositions of TiO2-D and TiO2-ND are significantly different. Thus, it is also evident that the O2 evolution rate on TiO2 is not affected by crystalline phases as reported previously.35
Increasing the calcination temperature from 200 °C to 800 °C leads to an improvement of O2 evolution on the TiO2-D and TiO2-ND samples. The crystallinity of TiO2-D is nearly constant and thus the change in the O2 evolution rate depending on calcination temperature is not attributable to the change in the crystallinity. However, calcination at 900 °C caused a reduction in the O2 evolution rate for both TiO2-D and TiO2-ND and the reason will be discussed later.
Relationship between O2 evolution rate and crystallite size
To evaluate the effect of crystallite size of TiO2 on the O2 evolution rate, we estimated average crystallite size (Dav) by a weighted average using the following equation:| Dav = DAXA/100 + DRXR/100 + DBXB/100 | (4) |
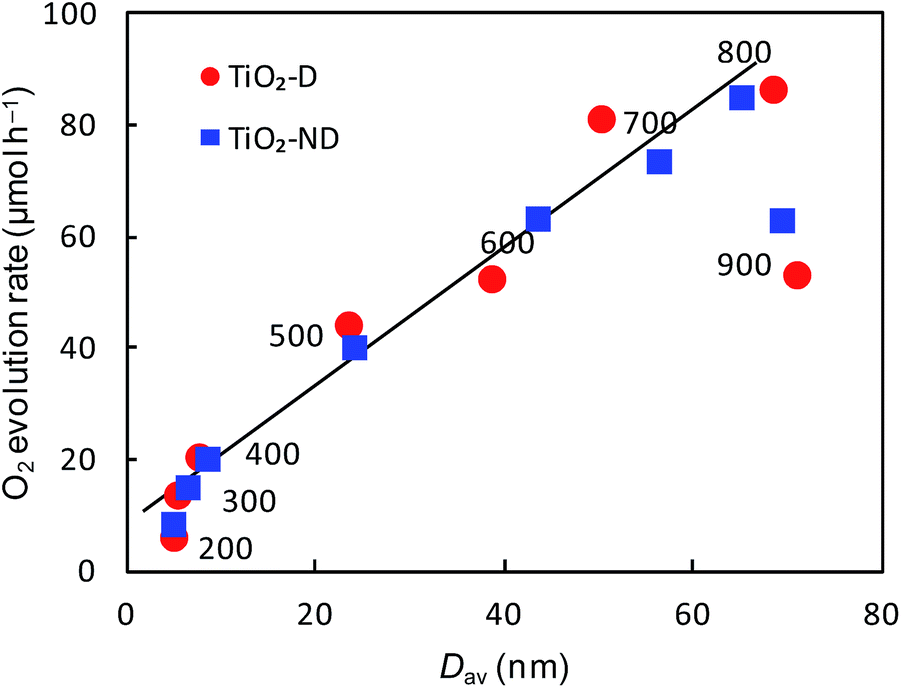 | ||
| Fig. 5 Relationships between Dav and O2 evolution rate on TiO2-D and TiO2-ND calcined at 200–900 °C for 2 hours. | ||
The calcination of TiO2-D and TiO2-ND at 900 °C reduces the O2 evolution rate significantly, although the crystallite sizes of them are slightly larger than those calcined at 800 °C. We have found that the crystallite size tends to approach a limiting value on increasing the calcination temperature and it remains almost constant at 900 °C regardless of the synthetic condition (i.e., with or without dialysis) and the calcination time. The SEM images in Fig. 6(a–c) show that no significant change in the morphology of TiO2-ND is observed when being calcined at 900 °C for 2, 3 and 4 h. Their crystallite sizes evaluated from the rutile (110) peaks in the XRD patterns are almost the same at 70.15 ± 0.54 nm. On the other hand, the O2 evolution rate clearly decreases with an increase in the calcination time (Fig. 6(d)). Maeda has reported20 that the highest O2 evolution was obtained by calcining commercially available rutile TiO2 at 800 °C in the presence of NaIO3 or FeCl3 as an electron acceptor. He ascribed the lower activity obtained at a calcination temperature higher than 800 °C to a decrease in the density of oxygen vacancies in TiO2. Amano et al.48 demonstrated the high photocatalytic activity of rutile TiO2 by creating oxygen vacancies through H2 reduction treatment at 700 °C. An increase in the density of oxygen vacancies leads to an increase in the donor density in semiconductors, which promotes the migration of the photogenerated carriers to enhance the photocatalytic activity. Therefore, the observed reduction in the O2 evolution rates on TiO2-D and TiO2-ND calcined at 900 °C is attributable to microscopic changes such as a decrease in the density of oxygen vacancies. Since the crystallite size is not increased by calcination at 900 °C for longer time, excess thermal energy might be used for decreasing the oxygen vacancies instead of growing the rutile particles.
 | ||
| Fig. 6 SEM images of TiO2-ND calcined at 900 °C for (a) 2, (b) 3, and (c) 4 hours and (d) effect of the calcination time on the O2 evolution rate and the crystallite size. | ||
Charge separation on polycrystalline TiO2 surface
Recently, improvement of photocatalytic activity by spatial charge separation has been reported for shape controlled or highly crystallized inorganic semiconductor photocatalysts.39,40,49–52 In such materials, photogenerated electrons and holes are transferred to the different crystal facets due to built-in electric field in the space-charge layer of them53 and thus the recombination of electrons and holes is suppressed. However, charge separation depending on crystal facets has been rarely reported for polycrystalline photocatalysts.Fig. 7(a) shows a TEM image of TiO2-ND after being used for the photocatalytic reduction of [PtCl6]2− in the presence of ethanol, indicating Pt deposition on specific particle surfaces. The EDS mapping is shown in Fig. 7(b) and the localized Pt deposition is confirmed. On the obtained Pt-deposited TiO2, Pb2+ ions were photocatalytically oxidized into PbO2. As shown in Fig. 7(c) and corresponding EDS mapping (Fig. 7(d)), the TEM images indicate that Pt and PbO2 are deposited on different crystal facets. The Pt or PbO2 loaded facets act as reduction or oxidation sites, respectively. This finding indicates the separation of these redox sites depending on the crystal facets of polycrystalline TiO2. Selected area electron diffraction patterns of the crystal facets deposited by Pt showed an inter-planar distance of 0.328 nm, corresponding to the (110) planes of the rutile phase (d = 0.3248 nm54). Ohno et al.39 reported that the reduction and the oxidation sites on the rutile TiO2 particles were on the (110) and the (011) faces, respectively.
 | ||
| Fig. 7 TEM images of (a) Pt loaded and (c) Pt and PbO2 co-loaded TiO2-ND which had been calcined at 800 °C for 2 hours. Element mappings of (b) Pt loaded and (d) Pt and PbO2 co-loaded TiO2-ND which had been calcined at 800 °C for 2 hours. | ||
Murakami et al.55 reported that the photocatalytic activity of decahedral anatase TiO2 for decomposition of acetaldehyde increased with an increase in the particle size up to ca. 40 nm and attributed it to the enhancement of the spatial separation of the oxidation sites and reduction sites. Small particle size is thought to be insufficient for efficient separation of the redox sites. The observed improvements of the O2 evolution activity of TiO2-D and TiO2-ND with an increase in the crystallite size are also attributable to the enhancement of the spatial charge separation.
Conclusions
Polycrystalline TiO2 particles were synthesized by the sol–gel method with and without dialysis procedures followed by calcination at 200–900 °C. The influence of their physicochemical properties on the photocatalytic activity for water oxidation has been investigated. No significant effects of the specific surface area and the crystalline phase on the O2 evolution rate were observed. A linear correlation between the crystallite size and the O2 evolution rate was obtained, which is attributed to the efficient spatial separation of the photogenerated carriers in large particles. The highest activity for O2 evolution was obtained on TiO2 calcined at 800 °C. The remarkable decrease in the photocatalytic activity of TiO2 calcined at 900 °C is likely due to microscopic changes such as the decrease in the density of surface oxygen vacancies, which suppresses the migration of the photogenerated carriers to be separated. Therefore, large particles where the spatial charge separation and charge transfer are efficiently achieved are required for designing an excellent photocatalyst for water splitting.Acknowledgements
We thank Prof. B. Ohtani at Hokkaido University and Prof. Y. Sakata at Yamaguchi University for useful discussions. We also thank Assoc. Prof. M. Yoshimoto and Mr Y. Fujiwara at Yamaguchi University for the zeta potential measurement. XRD measurements were performed at the Center for Instrumental Analysis, Yamaguchi University. SEM, TEM and TG measurements were carried out at the Innovation Center, Yamaguchi University. We thank Mr N. Takada and Mr H. Uejyo for their help in the measurements of TEM and selected area electron diffraction.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- S. Sato and J. M. White, Chem. Phys. Lett., 1980, 72, 83 CrossRef CAS.
- A. Tabata, N. Nishida, Y. Masaki and K. Tabata, Catal. Lett., 1995, 34, 245 CrossRef.
- H. Arakawa and K. Sayama, Catal. Surv. Jpn., 2000, 4, 75 CrossRef CAS.
- K. Maeda, Chem. Commun., 2013, 49, 8404 RSC.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 543 RSC.
- K. Domen, A. Kudo, T. Onishi, N. Kosugi and H. Kuroda, J. Phys. Chem., 1986, 90, 292 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992 CrossRef CAS.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858 CrossRef CAS PubMed.
- S. S. K. Ma, K. Maeda, R. Abe and K. Domen, Energy Environ. Sci., 2012, 5, 8390 CAS.
- A. Tanaka, K. Hashimoto and H. Kominami, J. Am. Chem. Soc., 2014, 136, 586 CrossRef CAS PubMed.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CrossRef CAS PubMed.
- Y. Sakata, Y. Matsuda, T. Nakagawa, R. Yasunaga, H. Imamura and K. Teramura, ChemSusChem, 2011, 4, 181 CAS.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CAS.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885 CrossRef CAS PubMed.
- H. Kominami, S. Murakami, J. Kato, Y. Kera and B. Ohtani, J. Phys. Chem. B, 2002, 106, 10501 CrossRef CAS.
- K. Maeda, ACS Catal., 2014, 4, 1632 CrossRef CAS.
- S. M. Thalluri, C. M. Suarez, M. Hussain, S. Hernandez, A. Virga, G. Saracco and N. Russo, Ind. Eng. Chem. Res., 2013, 52, 17414 CrossRef CAS.
- S. Yamazaki, Y. Fujiwara, S. Yabuno, K. Adachi and K. Honda, Appl. Catal., B, 2012, 121–122, 148 CrossRef CAS.
- X.-J. Dai, Y.-S. Luo, W.-D. Zhang and S.-Y. Fu, Dalton Trans., 2010, 39, 3426 RSC.
- G. Tian, H. Fu, L. Jing, B. Xin and K. Pan, J. Phys. Chem. C, 2008, 112, 3083 CAS.
- J. H. Pan, C. Shen, I. Invanova, N. Zhou, X. Wang, W. C. Tan, Q.-H. Xu, D. W. Bahnemann and Q. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 14859 CAS.
- K. Maeda, K. Teramura, T. Takata, M. Hara, N. Saito, K. Toda, Y. Inoue, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2005, 109, 20504 CrossRef CAS PubMed.
- X. Sum, K. Maeda, M. L. Faucheur, K. Teramura and K. Domen, Appl. Catal., A, 2007, 327, 114 CrossRef.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459 CrossRef CAS.
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, J. Phys. Chem. B, 2001, 105, 2417 CrossRef CAS.
- R. Abe, K. Sayama, K. Domen and H. Arakawa, Chem. Phys. Lett., 2001, 344, 339 CrossRef CAS.
- Z. Ding, G. Q. Lu and P. F. Greenfield, J. Phys. Chem. B, 2000, 104, 4815 CrossRef CAS.
- Y. Sakatani, D. Grosso, L. Nicole, C. Boissière, G. J. d. A. A. Soler-Illia and C. Sanchez, J. Mater. Chem., 2006, 16, 77 RSC.
- S. C. Pillai, P. Periyat, R. George, D. E. McCormack, M. K. Seery, H. Hayden, J. Colreavy, D. Corr and S. J. Hinder, J. Phys. Chem. C, 2007, 111, 1605 CAS.
- O.-O. Prieto-Mahaney, N. Murakami, R. Abe and B. Ohtani, Chem. Lett., 2009, 38, 238 CrossRef CAS.
- Y. K. Kho, W. Y. Teoh, A. Iwase, L. Mädler, A. Kudo and R. Amal, ACS Appl. Mater. Interfaces, 2011, 3, 1997 CAS.
- B. Ohtani, Y. Ogawa and S. Nishimoto, J. Phys. Chem. B, 1997, 101, 3746 CrossRef CAS.
- S. Yamazaki-Nishida, K. J. Nagano, L. A. Phillips, S. Cervera-March and M. A. Anderson, J. Photochem. Photobiol., A, 1993, 70, 95 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167 RSC.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- J. Zhang, Q. Xu, M. Li, Z. Feng and C. Li, J. Phys. Chem. C, 2009, 113, 1698 CAS.
- H. Zhang and J. F. Banfield, Am. Mineral., 1999, 84, 528 CrossRef CAS.
- H. Zhang and J. F. Banfield, Chem. Mater., 2005, 17, 3421 CrossRef CAS.
- L. Cao, D. Chen, W. Li and R. A. Caruso, ACS Appl. Mater. Interfaces, 2014, 6, 13129 CAS.
- M. Inagaki, Y. Hirose, T. Matsunaga, T. Tsumura and M. Toyoda, Carbon, 2003, 41, 2619 CrossRef CAS.
- S. Qourzal, A. Assabbane and Y. Ait-Ichou, J. Photochem. Photobiol., A, 2004, 163, 317 CrossRef CAS.
- J.-G. Li and T. Ishigaki, Acta Mater., 2004, 52, 5143 CrossRef CAS.
- F. Amano, M. Nakata and E. Ishinaga, Chem. Lett., 2014, 43, 509 CrossRef CAS.
- G. Liu, J. C. Yu, G. Q. Luc and H.-M. Cheng, Chem. Commun., 2011, 47, 6763 RSC.
- E. Bae, N. Murakami and T. Ohno, Appl. Catal., A, 2010, 380, 48 CrossRef CAS.
- H. Lin, L. Li, M. Zhao, X. Huang, X. Chen, G. Li and R. Yu, J. Am. Chem. Soc., 2012, 134, 8328 CrossRef CAS PubMed.
- K. Soma, A. Iwase and A. Kudo, Catal. Lett., 2014, 144, 1962 CrossRef CAS.
- J. Zhu, F. Fan, R. Chen, H. An, Z. Feng and C. Li, Angew. Chem., Int. Ed., 2015, 54, 9111 CrossRef CAS PubMed.
- M. Epifani, T. C. Capilla, T. Andreu, J. Arbiol, J. Palma, J. R. Morante and R. Diaz, Energy Environ. Sci., 2012, 5, 7555 CAS.
- N. Murakami, S. Kawakami, T. Tsubota and T. Ohno, J. Mol. Catal. A: Chem., 2012, 358, 106 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06151f |
| This journal is © The Royal Society of Chemistry 2016 |