
Hydroxylated di- and tri-styrylbenzenes, a new class of antiplasmodial agents: discovery and mechanism of action†
Abstract
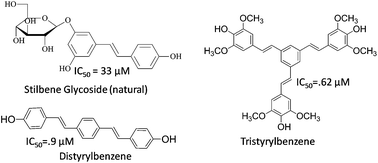
The first systematic evaluation of the antiplasmodial activity of the hydroxystilbene family of natural products and di/tristyrylbenzenes is described. A library of 27 diversely substituted hydroxy stilbenoids was rapidly synthesized using modified Knoevenagel–Perkin-decarboxylation–Heck sequences from readily available starting materials (i.e. hydroxybenzaldehyde–phenylacetic acid–arylhalide). These compounds were evaluated for in vitro antiplasmodial activity against three different strains of Plasmodium falciparum. Notably, 4,4′4′′-((1E,1′E,1′′E)-benzene-1,3,5-triyltris(ethene-2,1-diyl))tris(2,6-dimethoxyphenol) (27), an octupolar stilbenoid, showed IC50 (μM) values of 0.6, 0.5 and 1.36 while a distyrylbenzene (11) showed IC50 values of 0.9, 2.0 and 2.7 against 3D7 (chloroquine sensitive), Dd2 and Indo (chloroquine resistant) strains of Plasmodium falciparum respectively. Moreover, 27 and 11, which exhibited selectivity indices of 40 and >111 were also found to be nontoxic to the HeLa cell line. Microscopic studies revealed that the rings and trophozoites obtained from the 27 and 11 (an octupolar tristyrylbenzene and distyrylbenzene respectively) treated cultures were growth inhibited and morphologically deformed. These cultures also showed DNA fragmentation and loss of mitochondrial membrane potential (ΔΨm), suggestive of apoptotic death of the parasite. Together, these studies introduce di/tristyrylbenzenes as a new class of antimalarial agents.
 Please wait while we load your content...
Please wait while we load your content...

