
Preparation of halogenated furfurals as intermediates in the carbohydrates to biofuel process†
Abstract
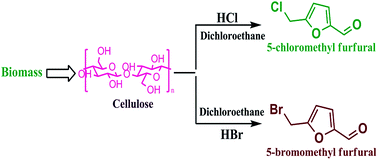
Lignocellulose derived halogenated furfurals are important chemicals that can serve as starting materials for diverse products such as drugs, polymers and fuels, including fuel additives. In this paper a protocol for the synthesis of 5-chloromethyl furfural (CMF) and 5-bromomethyl furfural (BMF) is put forward. The proposed process is based on a two liquid phases reaction composed of an aqueous hydrochloric or hydrobromic acid phase and 1,2-dichloroethane (DCE) organic phase. We have optimized and compared the yields of CMF and BMF in an open flask and in a closed reaction vessels. Utilization of the close reaction vessel resulted in not only higher yields in a shorter reaction time but also eliminated the necessity of the customary lithium salts additives previously advocated for this process. These additives were required in the open reaction systems in order to achieve reasonable yields. While a closed reaction vessel was previously reported for the production of CMF, and in this work its production was further improved, it is the first time a close vessel protocol for the production of BMF has been reported. In addition, improvement of the substrate to organic solvent ratio has been carried while yields of halogenated furfurals were maintained almost intact. NMR and UV-vis spectroscopy were used for identification and quantification of the products.
 Please wait while we load your content...
Please wait while we load your content...

