
Droplet-microarray on superhydrophobic–superhydrophilic patterns for high-throughput live cell screenings†
Abstract
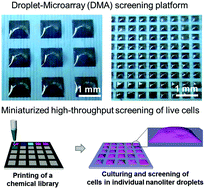
Cell-based high-content phenotypic screenings are widely used in fundamental research, pharmaceutical industry, and healthcare in order to simultaneously evaluate the effects of multiple compounds or gene overexpressions/knockdown on the phenotype of cells. Most screenings, particularly in the industrial sector, are performed using the microplate technology, which relies on high reagent and cell consumption, as well as on expensive liquid-handling robotics. Developing miniaturized screening platforms has been an important topic in the past decade. In this study, we demonstrate the applicability of the droplet-microarray platform based on superhydrophobic–superhydrophilic patterning for cell-based high-throughput screenings. We show the homogeneous seeding of cells and culturing of different adherent cell lines in individual droplets of different sizes. We demonstrate pipetting-free medium exchange, enabling cell culture in miniaturized droplet arrays for up to 5 days. We establish the methods of reverse transfection and reverse drug screening in individual nanoliter-sized droplets by printing transfection mixtures or drug molecules directly onto superhydrophilic spots prior to cell seeding.
 Please wait while we load your content...
Please wait while we load your content...

